
Αντιφώνηση Χρ. Καττάμη
Εισαγωγή
Σε ένα χρόνο συμπληρώνονται 80 χρόνια από την περιγραφή από τους Cooley και
Lee (1925), πέντε παιδιών με:
- Βαριά Αναιμία
- Ηπατοσπληνομεγαλία
- Οστικές παραμορφώσεις
- Έντονη μελάχρωση
- Λευκοκυττάρωση - Ερυθροβλαστοκυττάρωση.
Η ομάδα αυτή των αναιμιών απεδείχθη σαν το πιο συχνό μονογονιαδιακό νόσημα με
παγκόσμια διασπορά. Θα επιχειρήσω μια σύντομη ανασκόπηση της εντυπωσιακής διαχρονικής
εξέλιξης της μελέτης της παθογένειας και της αντιμετώπισης της νόσου.
Τρεις βασικές περίοδοι σηματοδοτούν αυτή την εξέλιξη :
Α. Περίοδος Πρώτη (Αρχική): 1925-1950
Χαρακτηρίζεται από τη συλλογή δεδομένων που οριοθέτησαν την κλινική και αιματολογική
ετερογένεια και τη γενετική βάση των Μεσογειακών Συνδρόμων (ΜΣ).
Β. Περίοδος Δεύτερη: 1950-1975
Ταυτίζεται με την εξέλιξη της βιοχημείας και της μελέτης της δομής των πρωτεϊνών.
Στην περίοδο αυτή εφαρμόσθηκαν νέες αιματολογικές και βιοχημικές μέθοδοι στη
διάγνωση και στη μελέτη της παθοφυσιολογίας της νόσου και τέθηκαν οι βάσεις
της θεραπείας και πιθανής πρόβλεψης.
Γ. Περίοδος Τρίτη:1975 μέχρι σήμερα
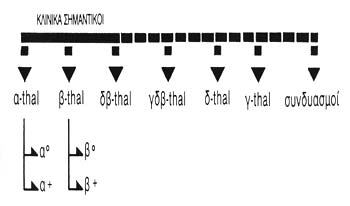
ΕΙΚΟΝΑ 1. Βασικοί τύποι μεσογειακής
αναιμίας.
Περίοδος
ΠΡΩΤΗ
Στην περίοδο αυτή, Αμερικανοί και Ευρωπαίοι, κυρίως Ιταλοί, αποτυπώνουν τα βασικά
κλινικά σημεία και αναζητούν την πιο πρόσφορη ονοματολογία της νόσου (Von Jackson
1889, Cooley and Lee 1925, Rietti 1925, Cooley et al 1927, Whipple and Bradford
1932, Michelli et al 1935, Valentine and Neel 1944).
Από το μεγάλο αριθμό των προτάσεων για την ονοματολογία επικράτησαν: α) στη
διεθνή βιβλιογραφία ο όρος " Τhalassemias" ή "Thalassemia Syndrome"
και β) στην ελληνική, ο όρος "Μεσογειακή Αναιμία" (ΜΑ) ή "Σύνδρομο
Μεσογειακής Αναιμίας" και οι πιο σπάνιοι όροι "Θαλασσαιμία" και
"Νόσος ή Αναιμία Cooley".
Από τις μελέτες της περιόδου αυτής αναγνωρίστηκαν:
Α) Η κλινική και εργαστηριακή ετερογένεια της νόσου και καθορίστηκαν οι βασικοί
κλινικοί φαινότυποι της μείζονος, της ενδιάμεσης και της ελάσσονος, ή minima
MA, όπως και του συνδυασμού μεσογειακής αναιμίας με τη δρεπανοκυτταρική.
Β) Ο οικογενής χαρακτήρας της νόσου. Από τις εργασίες του Καμινόπετρου (1939)
επιβεβαιώθηκε η μενδέλιος κληρονομικότητα υπολειπόμενου σωματικού τύπου. Η επιβεβαίωση
βασίστηκε στη μελέτη οικογενειών, στις οποίες και στους δύο γονείς ανευρίσκοντο
σταθερά αύξηση: της αντίστασης των ερυθρών, μικροκυττάρωση και υποχρωμία.
Γ) Η παρουσία της νόσου και σε άλλους πληθυσμούς εκτός των μεσογειακών.
Δ) Η αναποτελεσματικότητα της θεραπείας με σπληνεκτομή και μεμονωμένες μεταγγίσεις.
Στην περίοδο αυτή η εφαρμογή συχνών μεταγγίσεων ήταν αδύνατη, λόγω: α) συχνών
και σοβαρών επιπλοκών, β) αδυναμίας συντηρήσεως αίματος και γ) δυσκολίας εφαρμογής
της μετάγγισης, που απαιτούσε σύγχρονη χορήγηση από το δότη στον άρρωστο και
ειδικές συσκευές.
Για ιστορικούς λόγους αναφέρεται ότι η χρήση συντηρημένου αίματος εφαρμόζεται
μετά το Β΄ Παγκόσμιο Πόλεμο, οπότε άρχισαν να δημιουργούνται και οι πρώτες τράπεζες
αίματος στην Αμερική.
Περίοδος
Δεύτερη
Στη δεύτερη περίοδο, η εφαρμογή βελτιωμένων τεχνικών στη μελέτη της δομής των
πρωτεϊνών και της βιοσύνθεσης των πολυπεπτιδικών αλύσων, ως και η καθιέρωση
αξιόπιστων μεθόδων διάγνωσης, καθόρισαν τα κλινικά και εργαστηριακά χαρακτηριστικά
των αιματολογικών φαινοτύπων και προσδιόρισαν τους άξονες της πρόληψης και θεραπείας
των μεσογειακών συνδρομών.
Στην πρόοδο της περιόδου αυτής, η συμβολή της ελληνικής παιδιατρικής και ιδιαίτερα
της κλινικής μας, υπήρξε σημαντική και με αξιόλογη διεθνή αναγνώριση.
Ο προσδιορισμός της δομής της φυσιολογικής αιμοσφαιρίνης έδωσε το έναυσμα για
τη μελέτη της ΜΑ. Όπως γνωρίζετε, το μόριο της αιμοσφαιρίνης είναι τετραμερές
και αποτελείται από το πρωτεϊνικό κλάσμα και από 4 μόρια αίμης. Το πρωτεϊνικό
κλάσμα συνθέτουν δύο ζεύγη πολυπεπτιδικών αλύσων (πίνακας 1). Οι τρεις φυσιολογικές
αιμοσφαιρίνες Α, F και Α2 έχουν κοινό το ένα ζεύγος της α-αλύσου και διαφέρουν
στη δομή του δευτέρου ζεύγους, που είναι η β για την Α, η γ για την F και η
δ για την Α2. Στον ενήλικα επικρατεί η HbA, σε ποσοστά >95% (Lehmann and
Hunstman, 1974).
Από τις μελέτες που ακολούθησαν, διαπιστώθηκε ομάδα αναιμιών με ποιοτικές και
ποσοτικές διαταραχές στη σύνθεση των πολυπεπτιδικών αλύσων. Οι αναιμίες αυτές
χαρακτηρίστηκαν με το γενικό όρο αιμοσφαιρινοπάθειες. Από το σύνολο των αιμοσφαιρινοπαθειών
διαχωρίστηκαν τα σύνδρομα της μεσογειακής αναιμίας.
Πρόκειται για κλινικά σύνδρομα ετερογενή, που οφείλονται σε γενετικές διαταραχές,
οι οποίες οδηγούν σε μερική ή ολική καταστολή της σύνθεσης των πολυπεπτιδικών
αλύσων των φυσιολογικών αιμοσφαιρινών.
Με βάση την άλυσο της οποίας η σύνθεση διαταράσσεται, έχουν περιγραφεί διάφοροι
τύποι. Οι βασικοί τύποι μεσογειακής αναιμίας είναι η α, η β, η δβ, η γδβ και
η δ μεσογειακή αναιμία (εικόνα 1).
Κλινικό ενδιαφέρον παρουσιάζουν η α, η β, και η δβ μεσογειακή αναιμία που διαχωρίζονται
περαιτέρω σε δύο υποομάδες, την α0 και α+, και τη β0 και β+ μεσογειακή αναιμία.
Ο εκθέτης 0 υποδηλώνει την πλήρη αδυναμία σύνθεσης αλύσου, ενώ ο εκθέτης + τη
σύνθεση άλλοτε άλλης ποσότητας α ή β αλύσου, που υπολείπεται όμως της φυσιολογικής
(Καττάμης, 1989).
Η περίοδος αυτή χαρακτηρίζεται από συστηματικές και εκτεταμένες επιδημιολογικές
έρευνες για τη χαρτογράφηση της γεωγραφικής κατανομής των αιμοσφαιρινοπαθειών.
Αδρές εκτιμήσεις ανεβάζουν τον αριθμό των φορέων μεσογειακής και δρεπανοκυτταρικής
αναιμίας σε πάνω από 250 εκατομμύρια παγκοσμίως (WHO 1983).
Οι επιδημιολογικές μελέτες της γεωγραφικής κατανομής έδειξαν:
Α. Εντόπιση της ΜΑ σε πληθυσμούς της Μεσογείου, της εγγύς και μέσης Ανατολής,
της Ασίας, (ιδιαίτερα της νοτιοανατολικής), και της Β. Αφρικής.
Β. Πληθυσμιακές διαφορές στη συχνότητα και τη βαρύτητα της νόσου, όπως μεγαλύτερη
συχνότητα και βαρύτητα της κλινικής εικόνας της α-ΜΑ σε ασιατικούς πληθυσμούς
και μικρότερη συχνότητα με ηπιότερη βαρύτητα της β-ΜΑ στην Αφρική.
Γ. Εκλεκτική εντόπιση της δρεπανοκυτταρικής σε πληθυσμούς της Αφρικής, με μικρές
εστίες σε χώρες της Μεσογείου και της Μέσης Ανατολής, της HbE στην Ασία, και
της Ηb C στη Δυτική Αφρική.
Δ. Απουσία αιμοσφαιρινοπαθειών σε λαούς της Βόρειας, Δυτικής, Κεντρικής και
Ανατολικής Ευρώπης και της Αυστραλίας.
Ανάλογες επιδημιολογικές μελέτες επέτρεψαν την ακριβή χαρτογράφηση της γεωγραφικής
κατανομής των αιμοσφαιρινοπαθειών και στην Ελλάδα (Καττάμης, 1980). Όπως φαίνεται
από το χάρτη (εικόνα 2), υπάρχει μεγάλη ετερογένεια στη γεωγραφική κατανομή
των φορέων β-μεσογειακής αναιμίας. Παρατηρούνται περιοχές υψηλής συχνότητας
με ποσοστά >15 και 20% (όπως η Ρόδος, η Κύπρος, η Καρδίτσα κ.λπ.), περιοχές
μέσης συχνότητας 10-15% και χαμηλής <5%, όπως στη Βόρεια Ελλάδα.
Η δρεπανοκυτταρική αναιμία εντοπίζεται σε ορισμένες μόνο περιοχές, όπως της
Κωπαΐδας, της Χαλκιδικής, της Καρδίτσας και της Άρτας.
Η συμβολή της Α΄ Παιδιατρικής Κλινικής στις επιδημιολογικές αυτές μελέτες υπήρξε
ουσιαστική και πρωτοποριακή. Χαρακτηριστικό ήταν το διεθνές ενδιαφέρον ορισμένων
από τις πρώτες εργασίες, όπως η περιγραφή των πρώτων περιπτώσεων δρεπανοκυτταρικής
αναιμίας στον Ορχομενό, που προκάλεσε έντονες συζητήσεις για τον τρόπο εμφάνισης
της γενετικής διαταραχής σε άτομα της λευκής φυλής (Choremis et Zannos, 1956).
Τρεις άλλες μελέτες αφορούν στη συχνότητα των φορέων τριών γενετικών διαταραχών
των ερυθρών (ΜΑ, δρεπανοκυτταρικής και G6PDd) σε ελονοσόπληκτες περιοχές και
η θετική συσχέτιση της συχνότητας των διαταραχών με την ελονοσία. Τα ευρήματα
αυτά ενίσχυσαν την υπόθεση του ισορροπημένου πολυμορφισμού και συγκεκριμένα
της προστατευτικής δράσης έναντι της ελονοσίας που ασκούν στους φορείς, οι τρεις
οι ερυθροκυτταρικές διαταραχές (Choremis, Zannos, Kattamis 1962, Choremis et
al 1963, Fraser et al 1963).
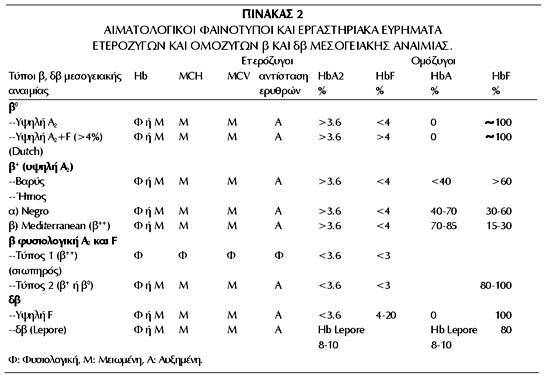
Στην ίδια περίοδο καθορίστηκαν οι αιματολογικοί φαινότυποι των ετεροζυγοτών
και των ομοζυγοτών της β και δβ μεσογειακής αναιμίας (πίνακας 2).
Στην Ελλάδα ανευρέθησαν όλοι οι φαινότυποι (Καττάμης 1989, 1995). Παράλληλα,
πρωτοπεριγράφηκε από την ομάδα μας ο αιματολογικός φαινότυπος της ετερόζυγου
β-μεσογειακής αναιμίας, με φυσιολογικές τιμές αιμοσφαιρίνων Α2 και F, που χαρακτηρίστηκε
ως "σιωπηλός". Ο φαινότυπος αυτός διαχωρίστηκε σε δύο υπότυπους, τον
ήπιο τύπο I, με φυσιολογικούς τους ερυθροκυτταρικούς δείκτες και ήπια κλινική
εικόνα με χαμηλή τιμή HbF στους διπλούς ετεροζυγότες και το βαρύ τύπο II με
παθολογικούς τους ερυθροκυτταρικούς δείκτες, με βαριά κλινική εικόνα και υψηλή
τιμή HbF στους αρρώστους (Καττάμης και συν, 1979). Οι δύο αυτοί φαινότυποι φορέων
β-ΜΑ είναι σχετικά συχνοί στην Ελλάδα και εύκολα μπορεί να διαφύγουν τη διάγνωση.
Ένα σημαντικό ποσοστό ετερόζυγων με τύπο II, απεδείχθη αργότερα ότι ήταν φορείς
της μετάλλαξης δβ Corfu. Πρόκειται για συνδυασμό απώλειας τμήματος του δ γόνου
με σημειακή μετάλλαξη στο β γόνο. Η μετάλλαξη περιγράφηκε από την ομάδα μας
και μέχρι σήμερα απαντάται μόνο στην Ελλάδα (Wainscoat et al 1985, Traeger -
Synodinos et al 1991).
Ανάλογη ταξινόμηση των αιματολογικών και κλινικών φαινοτύπων έγινε και για την
α-ΜΑ (πίνακας 3) (Καττάμης, 1989).
Η διευκρίνιση της παθοφυσιολογίας της β-ΜΑ δραστηριοποίησε την αναζήτηση τρόπων
αντιμετώπισής της. Η βασική διαταραχή αφορά στη μερική ή ολική αναστολή της
σύνθεσης της β αλυσίδας και της HbΑ, που οδηγεί σε βαριά αναιμία, με συνεπακόλουθα
τη σωματική καθυστέρηση, την ηπατοσπληνική διόγκωση και τις οστικές παραμορφώσεις.
Η άθροιση περίσσειας α-αλύσων επιδεινώνει την αναιμία με δύο μηχανισμούς: α)
με την κατακρήμνισή τους στους ερυθροβλάστες που οδηγεί στην καταστροφή τους
στο μυελό και σε μη αποδοτική ερυθροποίηση και β) με την κατακρήμνισή τους στα
ερυθρά που οδηγεί σε αιμόλυση (Weatherall and Clegg, 1981).
Η βασική αιτία της νόσου είναι η βαριά αναιμία. Ο Wolman, το 1964, πρότεινε
τις συχνές μεταγγίσεις για διατήρηση της αιμοσφαιρίνης σε ανεκτά όρια. Η κλινική
μας υιοθέτησε από πολύ νωρίς τη θεραπεία με συχνές μεταγγίσεις και δημοσίευσε
το 1970 τα πρόδρομα αποτελέσματα των ευεργετικών αποτελεσμάτων των μεταγγίσεων
στη σωματική αύξηση των αρρώστων. Χαρακτηριστική είναι η επιτάχυνση του ρυθμού
αύξησης μετά την έναρξη συχνών μεταγγίσεων, σε ένα άρρωστο που στα πρώτα χρόνια
διατηρείτο με χαμηλή Hb με περιστασιακές μεταγγίσεις. Η ασθενής, σήμερα 45 ετών,
έχει φυσιολογικό ύψος (εικόνα 3). Στην ίδια εργασία, φαίνεται ότι οι δείκτες
αύξησης των παιδιών με συχνές μεταγγίσεις και Ηb>8g/dl, κυμαίνονται στα φυσιολογικά
επίπεδα, ενώ των παιδιών που υπομεταγγίζοντο υπολείπονται σημαντικά (Κattamis
et al, 1970).
Πρόσφατες μελέτες της Κλινικής μας σε 405 αρρώστους ηλικίας 3-30 ετών, δίδουν
πιο ολοκληρωμένη εικόνα των επιπτώσεων των μεταγγίσεων στη σωματική ανάπτυξη
(εικόνα 4).
Ασθενείς σε συχνές μεταγγίσεις έχουν φυσιολογικό ρυθμό αύξησης μέχρι τα 10-12
χρόνια, ακολουθεί μια επιβράδυνση σε σχέση με τα φυσιολογικά άτομα στην εφηβεία
και στην αρχή της ενηλικίωσης. η αύξηση όμως συνεχίζεται και μετά από τα 20
χρόνια, έτσι ώστε μικρό σχετικά ποσοστό αρρώστων να βρίσκεται κάτω του φυσιολογικού
μετά το 20ο έτος (Kattamis et al, 1990).
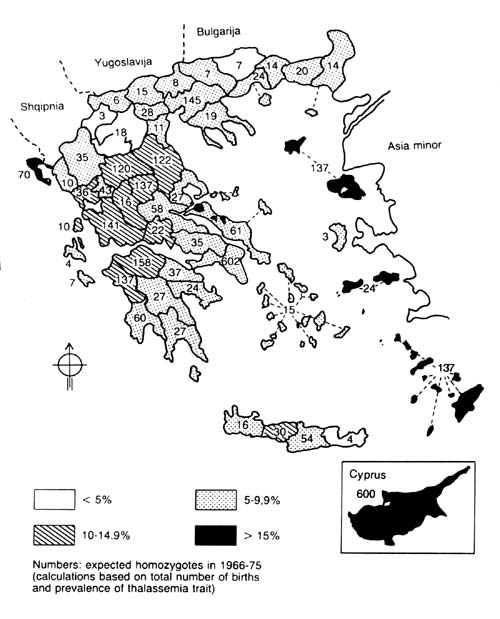
ΕΙΚΟΝΑ 2. Επιδημιολογία β
μεσογειακής αναιμίας από εκτεταμένες πληθυσμιακές μελέτες ανίχνευσης ετεροζυγοτών
στην
Ελλάδα (οι αριθμοί υποδηλούν αναμενόμενες γεννήσεις ομοζυγοτών, της δεκαετίας
1966-75, με βάση τον αριθμό
γεννήσεων και τη συχνότητα των ετεροζυγοτών στον πληθυσμό).

Τρίτη
Περίοδος
Η τρίτη περίοδος χαρακτηρίζεται από την ανάπτυξη της μοριακής ιατρικής και γενετικής
και την εφαρμογή συνεχώς βελτιούμενων τεχνικών για την ακριβή μελέτη της δομής
του DNA.
Το πρώτο επίτευγμα ήταν η χαρτογράφηση των γόνων των πολυπεπτιδικών αλύσων και
συγκεκριμένα των γόνων ε, γG, γA, δ, και β, του συμπλέγματος της β αλύσου στο
χρωμόσωμα 11, και των γόνων ζ, α2, α1 του συμπλέγματος της α-αλύσου στο χρωμόσωμα
16 (εικόνα 5).
Κατά την οντογονική εξέλιξη του ατόμου ενεργοποιούνται οι αντίστοιχοι με την
ηλικία γόνοι για τη σύνθεση των εμβρυονικών αιμοσφαιρινών (Gower 1, Portland,
Gower 2), των εμβρυϊκών HbF, και των αιμοσφαιρινών Α και Α2 του ενήλικα (Weatherall,
1991).
Στη συνέχεια, μοριακές μελέτες σε αρρώστους με ΜΑ έδειξαν ότι η αναιμία οφείλεται
σε βλάβες της δομής των γόνων των αλύσων της Hb. Οι δομικές αυτές βλάβες, σημειακές
ή ελλειμματικές, από απώλεια τμήματος ή και ολόκληρου του γόνου, οδηγούν σε
πλήρη ή μερική αναστολή της σύνθεσης της αντίστοιχης αλυσίδας.
Στη β ΜΑ, οι μεταλλάξεις είναι κατά κανόνα σημειακές, με σπάνιες εξαιρέσεις
τις ελλειμματικές, με απώλεια τμήματος του β γόνου. Αντίθετα, στην α μεσογειακή
επικρατούν οι ελλειμματικές μεταλλάξεις του ενός ή και των δύο α-γόνων. Το ίδιο
συμβαίνει και με τη δβ ΜA (Weatherall and Clegg, 1989).
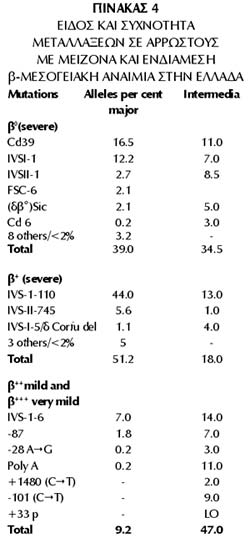
Μέχρι σήμερα έχουν περιγραφεί περίπου 200 μεταλλάξεις του β γόνου σε αρρώστους
με β μεσογειακή αναιμία. Ενδεικτικά απεικονίζεται μια σειρά μεταλλάξεων β ΜΑ
(Καττάμης, 1989). Οι σημειακές αυτές μεταλλάξεις εντοπίζονται από την αρχή μέχρι
και το τέλος της μεταγραφής και όχι μόνον στα εξόνια και εσόνια.
Ενδιαφέρον παρουσιάζει η χαρτογράφηση της επιδημιολογίας και πληθυσμιακής κατανομής
των μεταλλάξεων της β-ΜΑ.
Από τις μελέτες αυτές απεδείχθη: α) η παρουσία πληθυσμιακής εξειδίκευσης στις
μεταλλάξεις, β) 3-5 μεταλλάξεις σε κάθε πληθυσμό καλύπτουν το μεγαλύτερο ποσοστό
των παθολογικών γόνων, γ) η πληθυσμιακή διαφορά στο είδος των μεταλλάξεων ερμηνεύει
και την κλινική και την αιματολογική ετερογένεια που παρατηρείται μεταξύ πληθυσμών
και δ) σε πληθυσμούς της Μεσογείου παρατηρείται μεγαλύτερο φάσμα μεταλλάξεων
και διαφοροποίηση στη συχνότητα μεταλλάξεων. Στην ανατολική Μεσόγειο παρατηρείται
μια σαφής επικράτηση των β+ μεταλλάξεων, με συχνότερη τη IVS-I-110, ενώ στη
δυτική των β0 με προέχουσα τη CD39 (Weatherall et Clegg, 1996).
Στην Ελλάδα παρατηρήθηκε μια ιδιαιτερότητα στην επιδημιολογία και το φάσμα των
μεταλλάξεων (εικόνα 6). Μελέτες, κυρίως από τη δική μας ομάδα, έδειξαν την παρουσία
30 περίπου μεταλλάξεων β-ΜΑ. Οι πιο συχνές είναι οι β+ μεταλλάξεις (βαριές και
ήπιες) (Kattamis et al, 1990).
Ενδιαφέρον παρουσιάζει η μοριακή μελέτη του σιωπηρού αιματολογικού φαινοτύπου
σε 33 ετεροζυγότες, η οποία έδειξε τη συσχέτισή του με τρεις ήπιες, σχετικά
σπάνιες μεταλλάξεις του β γόνου, την +1480 (C"Τ), τη -101 (C"T), τη
+33 (C"G) και το συνδυασμό ετερόζυγης β-ΜΑ με τριπλασιασμένο α γόνο (Marangoudaki
et al, 1998).
Μελέτη της σχέσης γονότυπου με το κλινικό φαινότυπο σε πάνω από 500 αρρώστους
έδειξε μεγάλη συσχέτιση μεταξύ της βαρύτητας της κλινικής εικόνας και της βαρύτητας
της μετάλλαξης και του γονότυπου. Στους αρρώστους με ενδιάμεση μεσογειακή αναιμία
τουλάχιστον μία από τις δύο μεταλλάξεις ήταν ήπια. Μάλιστα, ορισμένες ήπιες
μεταλλάξεις βρέθηκαν μόνο σε αρρώστους με ήπια συμπτωματολογία. Αντίθετα, στους
αρρώστους με μείζονα μεσογειακή αναιμία, η παρουσία ήπιας μετάλλαξης είναι σπάνια
(πίνακας 4).
Στο σύνολο των αρρώστων αναγνωρίσθηκαν 75 διαφορετικοί γονότυποι με συνδυασμό
των πιο πάνω μεταλλάξεων.
Τα ευρήματα αυτά καταδεικνύουν την πρωταρχική σημασία του χαρακτηρισμού του
γονότυπου για τη διάγνωση, την πρόγνωση, τη θεραπεία και την πρόληψη. Για την
Κλινική μας, ο χαρακτηρισμός του γονότυπου αποτελεί προϋπόθεση ολοκληρωμένης
διάγνωσης και καθορισμού θεραπείας.
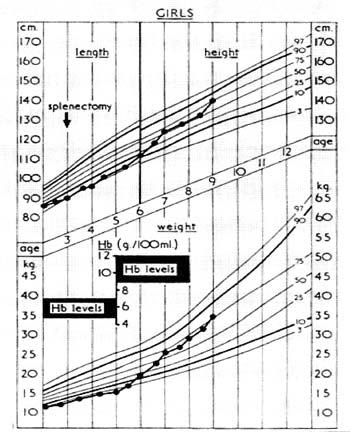
ΕΙΚΟΝΑ 3. Ρυθμός σωματικής
αύξησης ασθενούς προ και μετά την έναρξη θεραπείας
με συχνές μεταγγίσεις και διατήρηση Ηb σε επίπεδα 8.5 - 12g/dl.
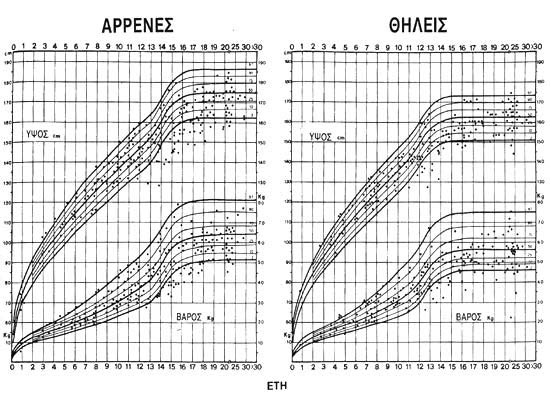
ΕΙΚΟΝΑ 4. Σωματική αύξηση
σε 405 αρρώστους με μεσογειακή αναιμία και συχνές μεταγγίσεις ηλικίας 3-30 ετών.
Πρόληψη
Η εφαρμογή μοριακών τεχνικών βοήθησε ουσιαστικά τη βελτίωση, καθώς και την αποτελεσματικότητα
της προγεννητικής διάγνωσης και της πρόληψης.
Η μελέτη του DNA επέτρεψε: α) την πρώιμη και έγκαιρη διάγνωση στην 11-12η εβδομάδα
κύησης, χρησιμοποιώντας ως υλικό DNA τον τροφοβλάστη, β) τον ακριβή και άμεσο
χαρακτηρισμό του γονότυπου για τη διάγνωση αρρώστου εμβρύου και γ) τη διακοπή
της κύησης σε αρχικά στάδια, χωρίς επιπτώσεις στη μητέρα.
Ο σύγχρονος αλγόριθμος των προγραμμάτων πρόληψης ΜΑ περιλαμβάνει: α) καθολικά
και αποτελεσματικά προγράμματα ανίχνευσης των ζευγαριών σε κίνδυνο, σε όλο τον
πληθυσμό, με αιματολογικές και βιοχημικές μεθόδους, β) χαρακτηρισμό των μεταλλάξεων
στους γονείς, γ) δυνατότητα προγεννητικής διάγνωσης και δ) διακοπή κύησης σε
διάγνωση αρρώστου εμβρύου (πίνακας 5).
Πρόσφατα προτείνεται, όταν υπάρχουν αδέλφια, συμπλήρωση του προγράμματος με
έλεγχο HLA ιστοσυμβατότητας αδελφών και εμβρύου, με στόχο: α) αν υπάρχει άρρωστος
συμβατός αδελφός, να γίνει συλλογή αίματος ομφάλιου λώρου για μεταμόσχευση και
β) σε περίπτωση αρρώστου εμβρύου, αν υπάρχει ιστοσυμβατός φυσιολογικός αδελφός,
προτείνεται ως εναλλακτική της διακοπής της κύησης, η μεταμόσχευση μυελού.
Σε προηγμένα υγειονομικά κράτη, η πρόληψη αποτελεί το βασικό σκέλος της αντιμετώπισης
της μεσογειακής αναιμίας. Όμως, η επιτυχία των προγραμμάτων εξαρτάται από πολλούς
παράγοντες, όπως αποδεικνύεται από τα αποτελέσματα των προγραμμάτων πρόληψης.
Στην Κύπρο, τη Σαρδηνία και την Ελλάδα τα προγράμματα πρόληψης έχουν μηδενίσει
ή ελαχιστοποιήσει τις γεννήσεις αρρώστων, ενώ στην Αγγλία, η μείωση γεννήσεων
αρρώστων είναι πολύ περιορισμένη (Bezis et al, 1995).
Αξιοσημείωτη πρόοδος στην πρόληψη χαρακτηρίζεται η δυνατότητα προεμφυτευτικής
διάγνωσης της ΜΑ και επιλογής για εμφύτευση φυσιολογικού εμβρύου. Στο Χωρέμειο
Ερευνητικό Εργαστήριο, το Τμήμα Κλινικής Γενετικής, με Διευθυντή τον Αναπληρωτή
Καθηγητή κ. Καναβάκη, πρωτοεφάρμοσε και πρωτοστατεί στην εφαρμογή της προεμφυτευτικής
διάγνωσης στην Ελλάδα (Kanavakis et al 1999, Traeger-Synodinos et al 2003).

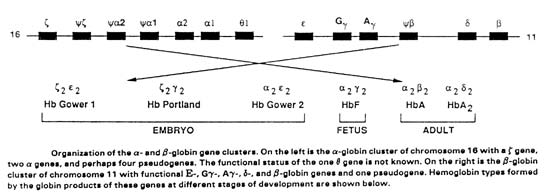
ΕΙΚΟΝΑ 5. Χαρτογράφηση γόνων
α και β συμπλέγματος φυσιολογικών αιμοσφαιρινών
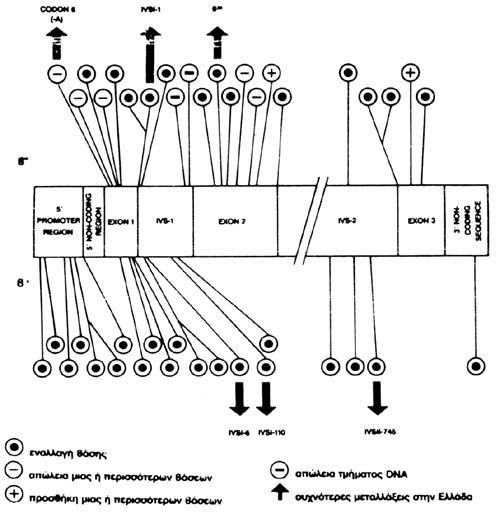
ΕΙΚΟΝΑ 6. Συχνές μεταλλάξεις
β-μεσογειακής αναιμίας. Με τόξα συχνότερες στην Ελλάδα.
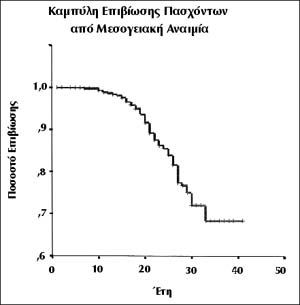
ΕΙΚΟΝΑ 7. Επιβίωση συνόλου
ασθενών της μονάδας ΜΑ της Α' Πανεπιστημιακής Κλινικής.
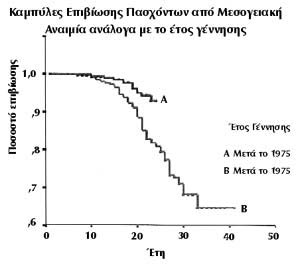
ΕΙΚΟΝΑ 8. Διαφορές επιβίωσης
σε δύο ομάδες ασθενών με βάση την ηλικία γέννησης (προ και μετά το 1975) και
την πληρότητα θεραπείας, κυρίως αποσιδήρωσης.
Θεραπευτικη
Αντιμετωπιση
Σημαντικές εξελίξεις σημειώθηκαν στη θεραπεία. Βελτιώθηκαν σημαντικά τα προγράμματα
μεταγγίσεων, κυρίως σε ό,τι αφορά στην ποιότητα του αίματος, στον τρόπο και
στις συσκευές μεταγγίσεων και την πρόληψη και αντιμετώπιση των παρενεργειών.
Ως βασικό παρασκεύασμα αίματος χρησιμοποιούνται τα πτωχά σε λευκά, συμπυκνωμένα
ερυθρά, που παρασκευάζονται με ειδικά φίλτρα ή με πλύση για την ελαχιστοποίηση
των πυρετικών αντιδράσεων. Για τον περιορισμό του αριθμού των μεταγγίσεων χορηγούνται
νεοκύτταρα και για τον περιορισμό της ισοανοποίησης, φαινοτυπικά συμβατό αίμα
(Καττάμης, 1989).
Στην περίοδο αυτή προστέθηκε συστηματικά στη θεραπεία η αποσιδήρωση και συμπληρωματικά
η πρόληψη και θεραπεία των ενδοκρινοπαθειών, των σοβαρών λοιμώξεων και των καρδιοπαθειών.
Η αποσιδήρωση άρχισε να εφαρμόζεται προοδευτικά από το 1975, με φάρμακο εκλογής
το Desferal, που χορηγείται σε ημερήσια υποδόριο έγχυση με τη βοήθεια ειδικής
αντλίας. Η μέθοδος, μολονότι αποτελεσματική, ήταν και είναι βασανιστική και
δυσκολοεφάρμοστη.
Η κλινική μας εφήρμοσε από νωρίς την αποσιδήρωση και μελέτησε την αποτελεσματικότητα
διαφόρων σχημάτων θεραπείας, με στόχο την ανεύρεση καθοδηγητικών δεικτών για
τον καθορισμό εξατομικευμένου αποτελεσματικού προγράμματος αποσιδήρωσης (Kattamis
et al 1979, Kattamis 1985).
Για το σκοπό αυτό μελέτησε και εφήρμοσε μεθόδους μελέτης ισοζυγίου σιδήρου και
μέτρησης των αποθεμάτων σιδήρου στο ήπαρ και στην καρδιά με μαγνητική τομογραφία.
Για την παράκαμψη των δυσκολιών και των μειονεκτημάτων της θεραπείας με Desferal
(DFO), παρασκευάσθηκαν και δοκιμάστηκαν διάφορες από του στόματος χηλικές ουσίες.
Από τις ουσίες αυτές, η Διφεριπρόνη (DFP) έχει εγκριθεί πρόσφατα ως χηλική ουσία
δεύτερης σειράς, λόγω της σχετικά περιορισμένης αποτελεσματικότητας και της
ακοκκιοκυτταραιμίας που μπορεί να προκαλέσει.
Πρόδρομες κλινικές μελέτες φάσης III, στις οποίες μετέχει ενεργά η κλινική μας,
έδειξαν ότι η ταυτόχρονη χορήγηση DFO και DFP έχει τόσο αθροιστική όσο και συνεργητική
δράση, όπως αποδεικνύεται από τη σύγκριση της ημερήσιας απέκκρισης σιδήρου με
μικτή θεραπεία, σε σχέση με τη μονοθεραπεία.
Με τη μικτή θεραπεία, η μέση ημερήσια απέκκριση σιδήρου αυξάνεται σημαντικά
και το ποσοστό των ασθενών με αρνητικό ισοζύγιο υπερβαίνει το 80%, έναντι μόλις
20% των ασθενών σε μονοθεραπεία με DFO ή DFP (Kattamis et al 2002, 2003).
Σε εξέλιξη βρίσκονται επίσης κλινικές μελέτες φάσης III, με ένα νέο από του
στόματος σκεύασμα, το ICL670.
Από τις εξελίξεις στην πρόληψη και τη θεραπεία των επιπλοκών σημειώνονται: α)
η πρόληψη της ηπατίτιδας Β, με τον έγκαιρο εμβολιασμό και τον έλεγχο των δοτών,
β) η σημαντική μείωση της μόλυνσης με τον ιό της ηπατίτιδας C με τον συνεχώς
βελτιούμενο έλεγχο των αιμοδοτών, γ) τα ικανοποιητικά αποτελέσματα θεραπείας
με την πεγκιλιωμένη IFN2α που διαπιστώσαμε πρόσφατα σε αρρώστους που δεν αντέδρασαν
στην απλή IFN, δ) ο μηδενισμός της ΗΙV λοίμωξης, ε) η ελαχιστοποίηση των πυρετικών
αντιδράσεων μετά την αφαίρεση των λευκών, και της ισοανοσοποίησης με το συστηματικό
φαινοτυπικό έλεγχο συμβατότητας αίματος και στ) η έγκαιρη διάγνωση και αντιμετώπιση
των διαταραχών της γλυκόζης και του υπογοναδισμού.
Στις εξελίξεις της θεραπείας θα σταθούμε για λίγο και στη γονιδιακή θεραπεία.
Παρά τις συστηματικές ερευνητικές προσπάθειες, η κλινική εφαρμογή αμιγούς γονιδιακής
θεραπείας στη ΜΑ φαίνεται απόμακρη. Δύο, όμως, μορφές θεραπείας που στηρίζονται
στη φιλοσοφία της γονιδιακής θεραπείας, έχουν ήδη εφαρμοσθεί. Η πρώτη αφορά
στην ενεργοποίηση της γ αλυσίδας και η δεύτερη στη μεταμόσχευση μυελού.
Για την ενεργοποίηση της γ αλύσου και αύξηση της HbF, χρησιμοποιούνται διάφορα
φάρμακα, κυρίως, όμως, η υδροξυουρία, η ερυθροποιητίνη και το οξυβουτυρικό οξύ.
Η σχετική αύξηση της HbF βελτιώνει την κλινική και αιματολογική εικόνα και κυρίως
τις θρομβωτικές κρίσεις αρρώστων με δρεπανοκυτταρική αναιμία. Ευεργετικά αποτελέσματα
παρατηρήθηκαν και σε αρρώστους με ορισμένους γονότυπους ενδιάμεσης μεσογειακής
αναιμίας.
Ριζική, όμως, θεραπεία επιτυγχάνεται με τη μεταμόσχευση μυελού των οστών, όπου
αντί της ενσωμάτωσης φυσιολογικού β γόνου στα αρχέγονα παθολογικά κύτταρα, όπως
επιδιώκεται με τη γονιδιακή θεραπεία, μεταμοσχεύονται αυτούσια φυσιολογικά αρχέγονα
κύτταρα από απόλυτα συμβατούς δότες. Η επιτυχία μετά από σωστή επιλογή ασθενών
φτάνει σήμερα το 90%. Ενδεικτική είναι η εμπειρία της Μονάδος Μεταμόσχευσης
Μυελού Οστών του νοσοκομείου μας, στην οποία έχουν μέχρι σήμερα μεταμοσχευτεί
67 παιδιά με μεσογειακή αναιμία. Μετά 5 χρόνια, η επιβίωση ανέρχεται στο 92%
και η πλήρης αποκατάσταση στο 89% (Peristeri et al 2000, 2002).
ΕπΙλογος
Όπως τονίσθηκε, η θεραπεία της ΜΑ με μεταγγίσεις ξεκίνησε πριν από 40 χρόνια
και έκτοτε βελτιώνεται προοδευτικά. Σήμερα, μπορούν ν' αποτιμηθούν τα μακροπρόθεσμα
αποτελέσματα της θεραπείας που συνοψίζονται σε:
- Ριζική θεραπεία, σε ένα σημαντικό ποσοστό αρρώστων (20%).
- Ελαχιστοποίηση των βασικών κλινικών συμπτωμάτων.
- Πρόληψη των επιπλοκών και αποτελεσματική αντιμετώπισή τους.
- Σημαντική βελτίωση της ποιότητας ζωής και της κοινωνικής προσαρμογής.
- Αύξηση του προσδόκιμου επιβίωσης.
Τα αποτελέσματα της μονάδας μας που αφορούν στην επιβίωση είναι ενδιαφέροντα
και συγκρίσιμα με τα διεθνή.
Όπως φαίνεται στην καμπύλη Kaplan-Mayer, στο σύνολο των ασθενών μας των 40 ετών,
η πιθανότητα επιβίωσης ανέρχεται στο 68% (εικόνα 7). Στην εικόνα 8 συγκρίνεται
η επιβίωση σε δύο ομάδες ασθενών, με βάση το έτος γέννησης.
Στην πρώτη ομάδα που γεννήθηκε προ του 1975 και άρχισε αργά την αποσιδήρωση,
η πιθανότητα επιβίωσης στα 25 χρόνια ανέρχεται στο 80% και στα 40 χρόνια στο
64%, ενώ στη νεώτερη δεύτερη ομάδα που γεννήθηκε μετά το 1975, η πιθανότητα
επιβίωσης στα 25 χρόνια είναι 95%. Οι δείκτες αυτοί υποδηλώνουν τη συνεχώς βελτιούμενη
αποτελεσματικότητα της θεραπείας και τα άριστα αποτελέσματα της μονάδας μας
συγκριτικά με άλλες (Λαδής, 2001).
Στο χρόνο που είχα στη διάθεσή μου προσπάθησα να σκιαγραφήσω τη διαχρονική εξέλιξη
των γνώσεων και των επιτευγμάτων στην αντιμετώπιση των μεσογειακών συνδρόμων,
υποσημαίνοντας ενδεικτικά ορισμένες από τις διεθνώς αναγνωρισμένες συμβολές
της κλινικής μας.
Αν πέτυχα το στόχο μου θα το κρίνετε εσείς. Εγώ θέλω να ευχαριστήσω πρώτα εσάς
που βρίσκεσθε σήμερα μαζί μας και τέλος, μα όχι λιγότερο, όλους τους συνεργάτες
μου, για τη μακροχρόνια και σημαντική προσφορά τους στην αντιμετώπιση των αρρώστων
με μεσογειακή αναιμία.
Σ' αυτή τη σύντομη ανασκόπηση, είναι έκδηλη η πολλαπλή συνεισφορά τους.
Ευχαριστίες
Ευχαριστώ πολύ τους στενούς συνεργάτες των κλινικοεργαστηριακών μελετών:
Α. Μεταξωτού - Μαυρομάτη, Β. Λαδή, Ε. Μπερδούση, Σ. Κωσταρίδου, Φ. Παλαμίδου,
Κ. Αθανασάκη, Α. Καττάμη, Π. Λαγό, Α. Καραμπούλα, Ε. Λαγκώνα, Θ. Λιακοπούλου.
Και των μοριακών ερευνών:
Ε. Καναβάκη, J. Treager - Συνοδινού, Μ. Τζέτη, Ε. Μαραγκουδάκη, Χρ. Βρεττού.
ΒΙΒΛΙΟΓΡΑΦΙΑ
eris PHh, Darbellay R, Entermann Ph. Prevention of β-Thalassemia major and Hb
Bart's Hydrops fetalis syndrome. Semin in Hematology 1995; 32:244.
Caminopetros J. Recherches sar Ι' anemie, erythroblastique infantile, des peuples
de la Mediterranee oriental. Am Med 1938; 43:27,104.
Cooley ΤΒ and Lee Ρ. Series of cases of splenomegaly in children with anemia
and peculiar bone changes. Tr Am Pediatr Soc 1925; 37:29.
Cooley ΤΒ, Witwer ER and Lee ΟΡ. Anemia in children with splenomegaly and peculiar
changes in the bones. Report of cases. Am J Dis Chi1192; 34:347.
Dameshek W. Target Cell Anemia. An erythroblastic type of Cooley's erythroblestic
anemia. Am J Med Sc 1940; 200:445.
Kanavakis Ε, Vrettou C, Palmer G, Tzetis Μ, Mastrominas, Μ, Traeger - Synodinos
J. Preimplantation genetic diagnosis in 10 couples at risk for transmitting
β-thalassemίa major: clinical experience including the initiation of six singleton
pregnancies. Prenat Diagn 1999; 19:1.217.
Kanavakis Ε, Traeger - Synodinos J. Pre-implantation genetic diagnosis in clinical
practice. J Med-Genet 2002; 39:6.
Kattamis C, Touliatos Ν, Haidas S, Matsaniotis Ν. Growth of children with thalassemia:
effect of different transfusion regimens. Arch Dis in Child 1970, 45:502.
Kattamis Α, Kassou C, Berdousi Η, Ladis V, Papasotiriou J, Kattamis C. Combined
Therapy with Desferrioxamine and deferiprone in thalassemic patients: effect
on urinary iron excretion. Blood 2003; 100:120a.
Kattamis Α, Kassou C, Ladis V, Berdousi Η, Alexopoulou Ε, Kaloumenou Ι, Kelekis
Ν, Kattamis C. Α 12-months trial of a combined Regimen of Deferiprone and Deferoxamine
in patients with Thalassemia. Hematologica 2003; 88:1.423.
Kattamis C, Metaxotou - Mavromati Α, Karamboula Α, Nasika Ε, Lehmann Η. The
clinical and hematological findings in children inheriting two types of thalassemia:
high Α2 type beta thalassemia and high F type of delta beta thalassemia. Br
J Haematol 1973; 2:29.
Kattamis C, Karamboula Κ, Metaxotou - Mavromati Α, Ladis V, Constantopoulou
Α. Prevalence of β0 and β+ thalassemia genes in Greek patients with homozygous
β-thalassemia. Hemoglobin 1979; 2:29.
Kattamis C, Metaxotou - Mavromati Α, Wood WG, Nash JR, Weatherall DJ. The heterogeneity
of normal HbA2 β-thalassemia in Greece. Br J Haematol 1979; 42:109.
Kattamis C, Lagos Ρ, Langona R. Chelation therapy and ferritin levels in patients
with homozygous β-thalassemia. Ιn: Progress in clinical and Biological Research.
Alan R-LISS Inc. New York 1979; 134:351.
Kattamis C. Screening for hemoglobinopathies. Ιn Bickel Η, Guthrie R, Hammarsen
G (eds) Neonatal screening for inborn errors of metabolism. Springer Berlin,
Heidelberg 1980; 133-147.
Kattamis C, Metaxotou - Mavromati Α, Ladis V, Tsiarta Η, Laskari S, Kanavakis
Ε. The clinical phenotype of β and δβ Thalassemias in Greece. Eur J Pediatr
1982; 139:135.
Kattamis C. Experience with desferrioxamine (Desferal) in thalassemic patients
in Greece. In. Hypertransfusion and iron chelation in thalassemia. Ed. Μ. Aksoy,
G. Birdwood, Huns Pub Berne, Stuttgart, Toronto 1985; 30.
Kattamis C, Liakopoulou Τ, Katamis Α. Growth and development in children with
thalassemia major. Acta Paediatr 1990; 36:111.
Kattamis C, Ηυ Η, Cheng G, Reese ΑΙ, Gonzalez - Redondo MJ, Kutlar Α, Kutlar
F, Huismann ΗΤ. Molecular characterization of β-thalassemia in 174 Greek patients
with thalassemia major. Br J Haematol 1990; 74:342.
Καττάμης Χρ. Θεραπευτική Αντιμετώπιση Μεσογειακής Αναιμίας. Μονογραφία 2η έκδοση,
Αθήνα 1989.
Καττάμης Χρ. Σύνδρομα μεσογειακής αναιμίας στο: Εφαρμογή μοριακών τεχνικών στη
μελέτη γενετικών νόσων, Καττάμης Χρ, Καναβάκης Ε (εκδότες). Α' Παιδιατρική Κλινική
Πανεπιστημίου Αθηνών, Αθήνα 1995; 26.
Λαδής Β. Σύγχρονη Θεραπευτική Αντιμετώπιση Μεσογειακής Αναιμίας. Βασικές αρχές
και αποτελεσματικότητα. Παιδιατρική 2001;64:285.
Lehmann Η, Hunstman RG. Man's Haemoglobins. North Holland Publ Com Amsterdam
1974.
Maragoudaki Ε, Vrettou C, Kanavakis Ε, Traeger - Synodinos J, Metaxotou - Mavromati
Α, Kattamis C. Molecular, hematological \ and clinical studies of a silent β
gene C~G mutation at Gbp3' to the termination codon (+1480 C~G) in twelve Greek
families. Br J Haematol 1998; 103: 45.
Michelli F, Penati F, Momigliano Levi G. Anemia ipocromica, splenomegalica con
ellitocitosi, poichilo-cistosi. Haematologica Archivio 1935; 16(1):5.
Rietti F. Ittero emolίtico primitivo. Atti Acad Scient Med Nat Ferrara 1925;
2:14.
Peristeri J, Kitra V, Goussetis Ε, Petropoulos D, Theodosaki, Kattamis Α, Graphakos
S. Haematopoietic stem cell transplantation for the management of hemo-globinopathies
in Greek patients. Tranf Science 2000; 23:263.
Peristeri J, Kitra V, Goussetis Ε, Pepropoulos D, Graphakos S. Hemopoietic stem
cell transplantation for thalassemic syndromes: the Greek experience. Bone Mar
Transpl 2002; 29(52):137.
Traeger - Synodinos J, Tzetis Μ, Kanavakis Ε, Metaxotou - Mavromati Α, Kattamis
C. Corfu δβ - thalassemίa mutation in Greece. Hematological phenotype and prevalence.
Br J Haematol 1991; 79:302.
Traeger - Synodinos J, Vrettou C, Palmer G, Tzetis Μ, Mastrominas Μ, Davies
S, Kanavakis Ε. Αn evaluation of preimplantation genetic diagnosis in clinical
genetic services through three years application for prevention of β - thalassemίa
major and sickle cell anemia. Μοl Hum Reprod 2003; 9:301.
Valentine WN and Neel JV. Hematologic and genetic study of the transmission
of thalassemia. Arch Int Med 1944; 74:185.
Von - Jaksch. Uber leukamies and leukocyte im kindesalter. Wien Κlin Wyhnschr
1989; 2:43/5.
Whipple GH, Bradfor WL. Racial and familial anemia of children associated with
foundamental disturbances of bone and pigment metabolίsm (Cooley - Von Jaksch).
Am J Dis Child 1932; 44:336.
Weatherall DJ and Clegg JB. The Thalassemias syndromes. 3rd Ed Blackwell Scientific
Publ Oxford, 1981.
Weatherall DJ. The New Genetics and Clinical Practice. 3rd Ed Oxford University
Press, 1991.
Weatherall D, Clegg J. Thalassemia; a global public health problem. Nature Medicine
1996; 2:847.
World Health Organization. Report on the community control of hereditary anemias.
Memorandum from a WHO meeting. Βυll World Health Org 1983; 61:63.
Wolman DJ. Transfusion therapy in Cooley's Anemia. Growth and health as related
to long range hemoglobin levels. Α progress report. Αnn Acad Sci 1964; 119:736.
