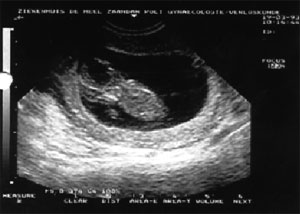
Eικόνα 1. Ινιακή εγκεφαλοκήλη
ΑΡΘΡΟ ΕΝΗΜΕΡΩΣΗΣ
Σύνδρομο Meckel-Gruber
Χρ. Ιαβάτσο
Ιατρός, Βοηθός
Γυναικολογικής Κλινικής ΒΟΥΓΙΟΥΚΛΑΚΕΙΟΥ Διαγνωστικού και Θεραπευτικού Κέντρου
Αλληλογραφία:
Χρ. Ρ. Ιαβάτσο
Σεϊζάνη 38, Ν. Ιωνία
14231 Αθήνα
Tηλ: 2102714896, 6948054119
Kατατέθηκε 12/11/2004
Εγκρίθηκε 10/12/04
Περίληψη
Το σύνδρομο Meckel-Gruber είναι ένα σπάνιο θανατηφόρο σύνδρομο. Κληρονομείται
κατά τον αυτόσωμο υπολειπόμενο τρόπο. Χαρακτηρίζεται από την τριάδα:
Α) Ινιακή εγκεφαλοκήλη
Β) Πολυκυστικοί νεφροί
Γ) Πολυδακτυλία
Τα έμβρυα ή τα νεογνά καταλήγουν λόγω υποπλασίας των πνευμόνων. Τα ποσοστά θνησιμότητας
είναι 100%. Η διάγνωση τίθεται υπερηχογραφικά ή με την αύξηση των επιπέδων της
a-FP του αμνιακού υγρού κατά την 11η ως 14η εβδομάδα κύησης. Συνιστάται η γενετική
καθοδήγηση των γονέων.
Όροι ευρετηρίου:
Ινιακή εγκεφαλοκήλη, πολυκυστικοί νεφροί, πολυδακτυλία.
ΕΙΣΑΓΩΓΗ
Το σύνδρομο Meckel-Gruber (MGS)(1-6) είναι ένα σπάνιο θανατηφόρο σύνδρομο που
κληρονομείται με βάση τους κανόνες της αυτοσωμικής υπολειπόμενης κληρονομικότητας
και εντοπίζεται στο χρωμόσωμα 17 (17q21-q24).
Χαρακτηρίζεται από την τριάδα:
1. ινιακή εγκεφαλοκήλη
2. μεγάλοι πολυκυστικοί νεφροί
3. πολυδακτυλία
Συνοδές ανωμαλίες(7) είναι:
1. Στοματικές σχιστίες
2. Ανωμαλίες γεννητικών οργάνων
3. Διαμαρτίες ΚΝΣ
4. Ίνωση ήπατος
Αιτία θανάτου είναι η υποπλασία των πνευμόνων. Με τη χρησιμοποίηση των υπερήχων
είναι εύκολη η προγεννητική διάγνωση στο δεύτερο τρίμηνο ή στο τέλος του πρώτου
τριμήνου της κύησης (11η ως 14η εβδομάδα κύησης).(8,9)
ΙΣΤΟΡΙΚΗ
ΑΝΑΔΡΟΜΗ
Ο Johan Friedrich Meckel(10) (1781-1833) ήταν ένας Γερμανός ανατόμος στο πανεπιστήμιο
του Halle. To 1822 περιέγραψε δύο αδέρφια με τις ακόλουθες διαμαρτίες:
1. Ινιακή μηνιγγοεγκεφαλοκήλη
2. Μικροκεφαλία
3. Υπερωιοσχιστία
4. Πολυκυστικούς νεφρούς
5. Πολυδακτυλία
Το 1934 ο G.B. Gruber(11) περιέγραψε ένα παρόμοιο σύνολο διαμαρτιών που πλέον
είναι γνωστό ως MGS. Όμως το σύνδρομο είχε ήδη περιγραφεί και τον 19ο αιώνα
από τους Vrolik(12) (1854), Foerster(13) (1862) και Casper(14) (1864). Το 1969
οι Opitz και Howe(15) διέκριναν τα κλινικά χαρακτηριστικά του συνδρόμου και
του έδωσαν την ονομασία (MGS). Το 1971 οι Mecke και Passarge πρότειναν ως τρόπο
κληρονομικότητας την αυτοσωμική υπολειπόμενη.
ΚΛΗΡΟΝΟΜΙΚΟΤΗΤΑ(16)
Το MGS κληρονομείται κατά τον αυτοσωμικό υπολειπόμενο τρόπο. Αυτό σημαίνει ότι
εμφανίζεται μόνο όταν το άτομο κληρονομήσει την ανωμαλία και από τους δύο γονείς-φορείς.
Ένα μοντέλο μίας τέτοιας οικογένειας είναι το ακόλουθο:
Aν Α είναι το φυσιολογικό γονίδιο και α το παθολογικό γονίδιο του MGS τότε θα έχουμε:
Γονείς: Αα (x) Aα
Φορέας Φορέας
Τέκνα: ΑΑ, Αα, Αα, αα
Φυσιολογικό Φορείς Πάσχον
25% 50% 25%
Η πιθανότητα εμφάνισης είναι πάντα η ίδια σε κάθε νέα εγκυμοσύνη ανεξαρτήτως φύλου. Οι τελευταίες έρευνες έδειξαν ότι το γονίδιο του MGS βρίσκεται στο χρωμόσωμα 17 (17q22-q23)(17,18) σε οικογένειες φινλανδικής καταγωγής ή στο χρωμόσωμα 11 (11q13)(19) σε οικογένειες της Μέσης Ανατολής και της Βορείου Αφρικής.
Eικόνα 1. Ινιακή εγκεφαλοκήλη
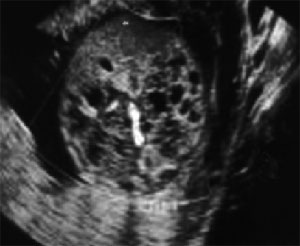
Eικόνα 2. Πολυκυστικοί νεφροί

Eικόνα 3. Πολυδακτυλία
ΣΥΧΝΟΤΗΤΑ(20)
Το MGS είναι ένα σπάνιο σύνδρομο με 200 αναφερόμενες περιπτώσεις. Επηρεάζει
με την ίδια συχνότητα άρρενα και θήλεα άτομα. Mε μεγαλύτερη συχνότητα παρουσιάζεται
στη Φινλανδία, τη Βόρεια Αφρική, το Βέλγιο την Ινδία, καθώς και σε συγκεκριμένες
υποομάδες πληθυσμού όπως οι Iνδιάνοι οι Gugarati, οι Bεδουίνοι, οι Τατάροι και
οι Εβραίοι Ashkenazi.
Παγκοσμίως εμφανίζεται με συχνότητα 1:13250 ως 1:140000 γεννήσεις. Όμως, στις
φινλανδικές οικογένειες είναι 1:8500.
Eνδεικτικά, οι συχνότητες του συνδρόμου σε διάφορες χώρες είναι:
Μεγάλη Βρετανία: 1:130000
Εβραίοι Ashkenazi: 1:50000
Φινλανδία: 1:8500
Μασαχουσέτη: 1:13250
Βέλγιο: 1:3000
Ινδιάνοι Gujarati: 1:1300
ΠΑΘΟΛΟΓΙΚΗ
ΦΥΣΙΟΛΟΓΙΑ
Υποστηρίζεται ότι το MGS προκαλείται από την ανώμαλη ανάπτυξη του μεσοδέρματος
που επηρεάζεται από αυξητικούς παράγοντες, γονίδια homeobox και γονίδια paired
domain.
ΙΣΤΟΛΟΓΙΚΑ
ΕΥΡΗΜΑΤΑ(21)
Η πρωταρχική ανωμαλία είναι η αποτυχία ανάπτυξης του ουρητήρα και του νεφρικού
στρώματος. Για αυτό τον λόγο οι νεφροί δείχνουν μικρή επινεφριδιακή διαφοροποίηση
και οι νεφρώνες ανευρίσκονται διογκωμένοι. Επίσης, σε όλο το νεφρικό παρέγχυμα
παρουσιάζονται λεπτοτοιχωματικές κύστεις.
Οι ηπατικές βλάβες ανευρίσκονται μόνο ως νεκροτομικά ευρήματα. Τέτοιες βλάβες
είναι:
α) αντιδραστικός πολλαπλασιασμός του χοληφόρου συστήματος
β) διάταση του χοληφόρου συστήματος
γ) πυλαία ίνωση
δ) εξάλειψη του πυλαίου φλεβικού συστήματος
ΦΥΣΙΚΗ
ΙΣΤΟΡΙΑ ΤΟΥ ΣΥΝΔΡΟΜΟΥ
Το σύνδρομο χαρακτηρίζεται από δυσπλαστικούς νεφρούς, γεγονός που οδηγεί το
έμβρυο σε ολιγοϋδράμνιο. Η υποπλασία των πνευμόνων αποτελεί τη βασική αιτία
θανάτου είτε ενδομητρικώς είτε αμέσως μετά τον τοκετό.
Περίπου το 1/3 των περιπτώσεων γεννιούνται νεκρά, ενώ τα 2/3 των νεογνών επιβιώνουν
το μέγιστο για 2 ώρες και 30 λεπτά. Σπάνια επιβιώνουν περισσότερο από 4 ημέρες
αν και έχουν αναφερθεί περιστατικά που επιβίωσαν από 5 μήνες ως και 4 χρόνια.
ΚΛΙΝΙΚΑ
ΧΑΡΑΚΤΗΡΙΣΤΙΚΑ ΤΟΥ ΣΥΝΔΡΟΜΟΥ
1. Ινιακή εγκεφαλοκήλη(22)
Χαρακτηρίζεται από προβολή ή πρόπτωση των στοιχείων του ρομβοειδούς πυρήνα,
του σκώληκα, της παρεγκεφαλίδας, της τρίτης και της τέταρτης κοιλίας του εγκεφάλου
διαμέσω της διευρυμένης οπίσθιας πηγής του κρανίου. Περιστασιακά, στο σάκο της
κήλης που δημιουργείται από τη διευρυμένη 3η κοιλία περιλαμβάνεται και ο μέσος
ινιακός φλοιός του εγκεφάλου. Τα προβαλλόμενα στοιχεία του ΚΝΣ καλύπτονται από
το σάκο της σκληράς μήνιγγας.
2. Πολυκυστικοί νεφροί(23)
Το πιο συχνό εύρημα του ΜGS είναι η κυστική δυσπλασία των νεφρών. Οι νεφροί
μεγεθύνονται 10-20 φορές. Οι ανώμαλοι αυτοί νεφροί λειτουργούν ελλιπώς και προκαλούν
ολιγοϋδράμνιο.
3. Πολυδακτυλία
Αφορούν τα άνω και τα κάτω άκρα. Υπάρχουν όμως και αναφερόμενες περιπτώσεις
GMS χωρίς πολυδακτυλία.
4. Ηπατική δυσγενεσία
Συχνά αναφέρεται ηπατική βλάβη. Παρουσιάζεται ποικιλομορφία του ενδοηπατικού
χοληφόρου συστήματος που χαρακτηρίζεται από:
α) αντιδραστικό πολλαπλασιασμό του χοληφόρου συστήματος
β) διάταση του χοληφόρου συστήματος
γ) πυλαία ίνωση
δ) εξάλειψη του πυλαίου φλεβικού συστήματος
5. Στοματικές σχιστίες
Συχνά συνυπάρχουν λυκόστομα και λαγώχειλος.
6. Ανωμαλίες των γεννητικών
οργάνων
Συνυπάρχει δυσκολία καθορισμού του φύλου του εμβρύου ή του νεογνού μέσω των
έξω γεννητικών οργάνων χωρίς χρωμοσωμική ανάλυση.
7. Σύνδρομο Dandy-Walker
Xαρακτηρίζεται από:
α) Κεντρική κύστη επικοινωνούσα με την 4η κοιλία
β) Αγενεσία του σκώληκα της παρεγκεφαλίδας
γ) Διαχωρισμό των παρεγκεφαλιδικών ημισφαιρίων
δ) Υδροκεφαλία
ΔΙΑΓΝΩΣΗ(24,25)
Η προγεννητική διάγνωση προκύπτει:
Α. Υπερηχογραφικώς:(26-28)
Είναι η καλύτερη μέθοδος για τη διάγνωση του συνδρόμου κατά την 11η έως τη 14η
εβδομάδα. Στο δεύτερο τρίμηνο η διάγνωση γίνεται δυσκολότερη, γιατί το ολιγοϋδράμνιο
μειώνει τη διακριτική ικανότητα των υπερήχων. Αρχικά καταγράφεται ινιακή εγκεφαλοκήλη
στο τέλος του πρώτου τριμήνου. Μέρος του εγκεφάλου και των μηνίγγων προπίπτει.
Επίσης, ανευρίσκονται μεγάλοι πολυκυστικοί νεφροί. Στο δεύτερο τρίμηνο ανευρίσκεται
πολυδακτυλία, αν δεν μας εμποδίσει το ολιγοϋδράμνιο. Οι πολυκυστικοί νεφροί
παρουσιάζονται στο 95-100% των περιπτώσεων του συνδρόμου. Στους νεφρούς αρχικά
ανευρίσκονται μικροσκοπικές κύστεις που καταστρέφουν το νεφρικό παρέγχυμα και
μεγεθύνουν το νεφρό κατά 10-20 φορές. Η ινιακή εγκεφαλοκήλη είναι παρούσα στο
60-80% των εμβρύων. Η πολυδακτυλία εμφανίζεται στο 55-75% των εμβρύων. Όταν
στον υπερηχογραφικό έλεγχο ανευρίσκονται τα δύο εκ των τριών χαρακτηριστικών
της τριάδας του συνδρόμου, διαγιγνώσκεται το MGS.
Β. Αυξημένα επίπεδα a-FP
στο αμνιακό υγρό μετά από αμνιοπαρακέντηση
Η a-FP του αμνιακού υγρού ανευρίσκεται αυξημένη κατά την 14η-16η εβδομάδα.
Γ. Μαγνητική τομογραφία
(MRI)
Χρησιμοποιείται συμπληρωματικώς του υπερηχογραφήματος μετά το β' τρίμηνο ή σε
περιπτώσεις καθυστερημένης διάγνωσης, όταν έχουν ήδη διαπιστωθεί τα χαρακτηριστικά
του συνδρόμου. Κυρίως, χρησιμοποιείται σε περίπτωση ολιγοϋδραμνίου. Καταδεικνύει
το μέγεθος και τη μορφολογία των νεφρών καθώς και την εγκεφαλοκήλη.
ΔΙΑΦΟΡΙΚΗ
ΔΙΑΓΝΩΣΗ
Το σύνδρομο πρέπει να διαφοροδιαγνωσθεί από:
Α. Την τρισωμία 13 μέσω ελέγχου του καρυοτύπου, ο οποίος ανευρίσκεται φυσιολογικός
στο MGS.
B. Τη νόσο των πολυκυστικών νεφρών που κληρονομείται κατά τον αυτοσωμικό επικρατούντα
τρόπο.
ΠΡΟΓΝΩΣΗ
Πρόκειται για σπάνιο θανατηφόρο σύνδρομο. Το 1/3 των εμβρύων γεννιέται νεκρό,
ενώ τα 2/3 αυτών καταλήγουν λίγες ώρες ή ημέρες μετά τον τοκετό. Ελάχιστα έμβρυα
επιβιώνουν λίγους μήνες με κάκιστη ποιότητα ζωής. Ο θάνατος προκαλείται λόγω
της υποπλασίας των πνευμόνων. Το ποσοστό θνησιμότητας είναι 100%.
ΧΕΙΡΙΣΜΟΣ
Όταν υποπτευόμαστε το σύδρομο MGS, επιβάλλεται ο έλεγχος του καρυοτύπου για
τη διαφοροδιάγνωση από την τρισωμία 13. Η διάγνωση επιτυγχάνεται κατά την 14η
ως την 16η εβδομάδα της κύησης μέσω των υπερηχογραφικών ευρημάτων και των αυξημένων
επιπέδων a-FP στο αμνιακό υγρό. Για τον αποκλεισμό του συνδρόμου απαιτείται
παρακολούθηση της εγκύου έως την 20η εβδομάδα κύησης.
Η συμβουλή του γυναικολόγου είναι ο τερματισμός της κύησης. Αν οι γονείς επιθυμούν
τη συνέχισή της, το έμβρυο είναι δεδομένο ότι θα γεννηθεί νεκρό ή θα καταλήξει
λίγο μετά τη γέννησή του. Συνιστάται γενετική καθοδήγηση των γονέων για την
πιθανότητα επανεμφάνισης του συνδρόμου σε νέα κύηση (25%).
Summary
Iavazzo C.
Meckel-Gruber Syndrome.
Hellen Obstet Gynecol 17(1): 51-55, 2005
Meckel-Gruber syndrome is a rare lethal autosomal recessive condition which is characterized by the triad of occipital encephalocele, large polycystic kidneys and polydactyly. Fetal pulmonary hypoplasia is the cause of death. The mortality rate is 100%. A prenatal ultrasound and elevated amniotic fluid alpha fetoprotein at 11th to 14th week of pregnancy are the clues of the diagnosis. A geneticist should be consulted.
Key words: Occipital encephalocele, polycystic kidneys, polydactyly.
ΒΙΒΛΙΟΓΡΑΦΙΑ
1. Mecke S, Passarge E. Encephalocele, Polycystic Kidneys and Polydactyly as
an Autosomal Recessive Trait simulating certain other Disorders: The Meckel
Syndrome. Ann Genet 1971; 14(2):97-103.
2. Farag TI, Usha R, Mady St, Al-Naydy K, El-Badramany M. Phenotypic variation
in Meckel-Gruber syndrome. Clin Genet 1990; 38:176-179.
3. Jaffe R. Meckel-Gruber syndrome. Fetus 1991; 1:1-3.
4. Buyse ML (Ed.). Birth defects encyclopedia. Blackwell Scientific, 1990.
5. Altmann P, Wagenbichler P, Schaller AA. Casuistic report on the Gruber or
Meckel syndrome. Hum Genet 1977; 38:357-362.
6. Jones KL. Meckel-Gruber syndrome. Smith's Recognizable patterns of human
malformation, 5th ed. Philadelphia: W.B. Saunders, 1997.
7. Paavola P, Salonen R, Baumer A. Clinical and genetic heterogeneity in Meckel
syndrome. Hum Genet 1997; 101(1):88-92.
8. Braithwaite JM, Economides DL. First-trimester diagnosis of Meckel-Gruber
syndrome by trans-abdominal sonography in a low-risk case. Prenat Diagn 1995;
15(12):1168-1170.
9. Sepulveda W, Sebire NJ, Souka A, Snijders RJ, Nicolaides KH. Diagnosis of
the Meckel-Gruber syndrome at eleven to fourteen weeks' gestation. Am J Obstet
Gynecol 1997; 176(2):316-319.
10. Meckel JF. Beschreibung zweier, durch sehr ahn-liche Bildungsabweichungen
entstellter Gesch-wister. Dtsch Arch Physiol 1822; 7:99-172.
11. Gruber GB. Beitrage zur frage gekoppelter Miszbildungen (Akrocephalosyndactylie
und Dy-sencephalia splanchnocystica). Beitr Path Anat 1934; 93:459-476.
12. Vrolik W. Tabulae ad illustrandum embryogenesis hominis et mammalium, tam
naturalem quam abnormen. Tab. LX: Imperfecta maxillae inferioris conditio, T.O.
Weigel ed. Leipzig 1854.
13. Foerster A. Zur Casuistik der Hirnkrankheiten. Wurzb Med Zeitschr 1862;
3:193-210.
14. Casper JL. Eine Missgeburt selttenster Art. Lebensfahigkeit. Berl Klin Wschr
1864; 1:9-10.
15. Opitz J, Howe JJ. The Meckel Syndrome (Dyse-ncephalia splanchnocystica,
the Gruber syndrome) In: Congenital malformations Syndromes, Birth Defects:
Original Article Series, The National Foundation, New York 1969; 2:167-179.
16. Johnson CA, Gissen P, Sergi C. Molecular pathology and genetics of congenital
hepatorenal fibrocystic syndromes. J Med Genet 2003; 40(5):311-319.
17. Paavola P, Salonen R, Weissenbach J. The locus for Meckel syndrome with
multiple congenital anomalies maps to chromosome 17q21-q24. Nat Genet 1995;
11(2):213-215.
18. Paavola P, Avela K, Horelli-Kuitunen N. High-resolution physical and genetic
mapping of the critical region for Meckel syndrome and Mulibrey Nanism on chromosome
17q22-q23. Genome Res 1999; 9(3):267-276.
19. Roume J, Genin E, Cormier-Daine V. A gene for Meckel syndrome maps to chromosome
11q13. Am J Hum Genet 1998; 63(4):1095-1101.
20. Salonen R, Norio R. The Meckel syndrome in Finland:epidemiologic and genetic
aspects. Am J Med Genet 1984; 18(4):691-824.
21. Nyberg DA, Halley D, Mahony BS, Hirsch JH, Luthy DA, Hickok D. Meckel-Gruber
Syndrome: importance of prenatal diagnosis. J Ultrasound Med 1990; 9:691-696.
22. Salonen R. The Meckel syndrome: clinicopa-thological findings in 67 patients.
Am J Med Genet 1984; 18(4):671-689.
23. Ahdab-Barmada M, Claassen D. A distinctive triad of malformations of the
central nervous system in the Meckel-Gruber syndrome. J Neuropathol Exp Neurol
1990; 49(6):610-620.
24. Blankenberg TA, Ruebner BH, Ellis WG. Pathology of renal and hepatic anomalies
in Meckel syndrome. Am J Med Gen 1987; 3:395-410.
25. Nyberg DA, Halley D, Mahony BS, Hirsch JH, Luthy DA, Hickok D. Meckel-Gruber
syndrome: importance of prenatal diagnosis. J Ultrasound Med 1990; 9:691-696.
26. Dahiya N, Vijay S, Prabhakar S, Subramamam S, Dahiya Neha. Antenatal Ultrasound
Diagnosis of Meckel-Gruber Syndrome. Ind J Radiol Imag 2001; 11(4):199-201.
27. Herman TE, Siegel MJ. Special imaging casebook. Meckel-Gruber syndrome.
J Perinatol 1996; 16(2 Pt 1):144-146.
28. Yapar EG, Ekici E, Dogan M. Meckel-Gruber syndrome concomitant with Dandy-Walker
mal-formation: prenatal sonographic diagnosis in two cases. Clin Dysmorphol
1996; 5(4):357-362.
29. Ramadani HM, Nasrat HA. Prenatal diagnosis of recurrent Meckel syndrome.
Int J Gynecol Obstet 1992; 39:327-332.