Αμφοτερόπλευρη γοναδική αγενεσία
και παρουσία του γονιδίου TSPY:
Δύο νέα χαρακτηριστικά του συνδρόμου
Mayer - Rokitansky - Küster - Hauser
Ε. Πλευράκη1
Μ. Κήτα1
Δ.Γ. Γουλής1
Χ. Χατζησεβαστού-Λουκίδου2
Α.Φ. Λαμπρόπουλος3
Ε. Καζανζίδου4
Α. Αβραμίδης1
1Ενδοκρινολογική Κλινική, Γ.Ν. «Ιπποκράτειο», Θεσσαλονίκη
2Εργαστήριο Κυτταρογενετικής, Α' Παιδιατρική Κλινική Α.Π.Θ., Γ.Ν. «Ιπποκράτειο», Θεσσαλονίκη
3Εργαστήριο Γενικής Βιολογίας Α.Π.Θ.
4Ακτινολογικό Εργαστήριο, Γ.Ν. «Ιπποκράτειο», Θεσσαλονίκη
Αλληλογραφία: Ε. Πλευράκη Ενδοκρινολογική Κλινική Γ.Ν.Θ «Ιπποκράτειο» Κων/πόλεως 49 546 42 Θεσσαλονίκη Τηλ.: 2310-892.038
Κατατέθηκε: 8/2/2003, Εγκρίθηκε: 16/3/2003
Περίληψη
Σκοπός: Η περιγραφή των κλινικών και εργαστηριακών χαρακτηριστικών έξι ασθενών με σύνδρομο Mayer - Rokitansky - Kster - Hauser (MRKH).
Υλικό και Μέθοδοι: Μελετήσαμε 6 ασθενείς με ΜRKH, ηλικίας 19,2±1,8 ετών. Σε όλες τις ασθενείς έγινε βασικός ορμονικός έλεγχος της υπόφυσης, του θυρεοειδούς αδένα και των γονάδων. Το γεννητικό σύστημα μελετήθηκε με υπερηχογράφημα, μαγνητική τομογραφία και λαπαροσκόπηση, το ουροποιητικό με υπερηχογράφημα και ενδοφλέβια πυελογραφία, ενώ ο σκελετός εκτιμήθηκε με απλές ακτινογραφίες. Oι ασθενείς ελέγχθηκαν με χρωμοσωμική ανάλυση. Η παρουσία του TSPY γονιδίου του χρωμοσώματος Υ εκτιμήθηκε με PCR και nested PCR.
Αποτελέσματα: Όλες οι ασθενείς είχαν υποπλαστικό, τυφλό κόλπο. Τα δευτερογενή χαρακτηριστικά του φύλου και ο βασικός ορμονικός έλεγχος ήταν φυσιολογικά σε πέντε ασθενείς, ενώ σε μία διαπιστώθηκε υπεργοναδοτροφικός υπογοναδισμός και αμφοτερόπλευρη γοναδική αγενεσία. Όλες είχαν υποπλαστική μήτρα, ενώ σε μία οι πρόδρομοι σχηματισμοί της ήταν ασύμμετροι. Τέσσερις ασθενείς είχαν φυσιολογικούς ωαγωγούς και δύο ετερόπλευρη απλασία. Πέντε ασθενείς είχαν δυσπλαστικές βλάβες του σκελετού και τρεις των νεφρών. O καρυότυπος ήταν 46,ΧΧ σε όλες τις ασθενείς. Η PCR ήταν φυσιολογική, ενώ με την nested PCR αναγνωρίστηκε το TSPY γονίδιο σε 2 ασθενείς.
Συμπεράσματα: Το MRKH είναι δυνατό να συνυπάρχει με διαταραχές στη διάπλαση των γονάδων. Μια υποομάδα ασθενών φέρει το TSPY γονίδιο του Υ-χρωμοσώματος.
Όροι ευρετηρίου: Γοναδική αγενεσία, σύνδρομο Rokitansky, αγενεσία παραγώγων πόρου του Μüller, γοναδοβλάστωμα.
ΕΙΣΑΓΩΓΗ
Το σύνδρομο Mayer - Rokitansky - Küster - Hauser (MRKH), το οποίο αναφέρεται και ως απλασία των παραγώγων του πόρου του Müller, αναγνωρίστηκε για πρώτη φορά από τον Mayer το 1829.(1) To 1838 o Rokitansky το περιέγραψε ως αγενεσία της μήτρας και του κόλπου, που την απέδωσε σε διαταραχή της ανάπτυξης των παραγώγων του πόρου του Müller.(2) Το 1910 ο Küster διαπίστωσε τη συνύπαρξη ανωμαλιών στους νεφρούς(3) και το 1961 ο Hauser το διέκρινε από το σύνδρομο των θηλεοποιών όρχεων.(4)
Η συχνότητά του είναι 1:4.000-5.000 γεννήσεις θήλεων νεογνών και αποτελεί τη δεύτερη αιτία της πρωτοπαθούς αμηνόρροιας.(5) Χαρακτηρίζεται από δυσγενεσία των παραγώγων του πόρου του Müller, αλλά η ακριβής αιτιολογία του δεν είναι γνωστή μέχρι σήμερα.(5,6) Αφορά θήλεα άτομα με φυσιολογικό καρυότυπο, με φυσιολογική έκκριση γοναδοτροπινών, με φυσιολογικές στη λειτουργία τους ωοθήκες και με τη δυνατότητα επίτευξης ωοθυλακιορρηξίας.(7) Oι αναφορές της γοναδικής αγενεσίας είναι σπάνιες και συνήθως αφορούν άτομα με καρυότυπο 46,ΧΥ.(8,9)
Σκοπός της μελέτης ήταν η περιγραφή των κλινικών και των εργαστηριακών ευρημάτων των ασθενών με MRKH, καθώς και η διερεύνηση των πτυχών της αιτιολογίας του δια μέσου της γενετικής ανάλυσης.
ΥΛΙΚO ΚΑΙ ΜΕΘOΔOΙ
Στη μελέτη συμπεριλήφθηκαν έξι ασθενείς, μέσης ηλικίας 19.2 ±1.8 έτη (μέση τιμή ± SD), που προσήλθαν στην Ενδοκρινολογική Κλινική του Γ.Ν. Θεσσαλονίκης «Ιπποκράτειο» λόγω πρωτοπαθούς αμηνόρροιας, κατά τα έτη 2000-2001.
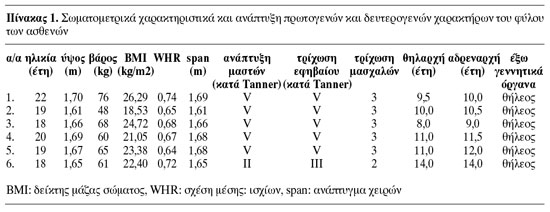
Ιστορικό και κλινική εξέταση: Στις ασθενείς εκτιμήθηκαν το ατομικό αναμνηστικό, το περιγεννητικό ιστορικό, η ύπαρξη άλλων θηλέων συγγενών με πρωτοπαθή αμηνόρροια, η ηλικία της μητέρας κατά την κύηση και η έκθεσή της σε τοξικούς παράγοντες. Στην κλινική εξέταση εκτιμήθηκαν τα σωματομετρικά χαρακτηριστικά (ύψος, βάρος, δείκτης μάζας σώματος (BMI), λόγος μέσης: ισχίων (WHR), ανάπτυγμα χειρών (span)), η ανάπτυξη των δευτερογενών χαρακτήρων του φύλου και η ανάπτυξη των έξω γεννητικών οργάνων κατά Tanner.
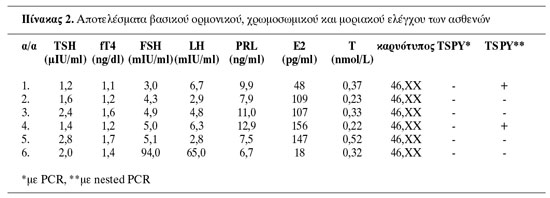
Oρμονικός και βιοχημικός έλεγχος: Στις ασθενείς προσδιορίσθηκαν η θυλακιοτρόπος ορμόνη (FSH) (φυσιολογικές τιμές ωοθυλακικής φάσης: 2,8-11,3mIU/ml), η ωχρινοτρόπος ορμόνη (LH) (ωοθυλακική φάση: 1,1-11,6mIU/ml), η προλακτίνη (PRL) (ωοθυλακική φάση: 4,5-33,0ng/ml), η οιστραδιόλη (Ε2) (ωοθυλακική φάση: 12-160pg/ml), η ολική τεστοστερόνη (Τ) (1,04-2,43nmol /L), η θυρεοτρόπος ορμόνη (TSH) (0,4-4,0μIU/ml) και η ελεύθερη θυροξίνη (fT4) (0,8-1,9ng/dl) με τη μέθοδο της ανοσοχημειοφωταύγειας (Ιmmunochemiluminescent assay - ILMA) στον αναλυτή Immulite 2000 DPC, Los Angeles, USA. Επίσης, προσδιορίσθηκαν η προγεστερόνη του ορού (ωχρινική περίοδος: 40-150pg/ml), η χοριακή γοναδοτροπίνη (hCG) (0-10mIU/ml) και η α-εμβρυϊκή πρωτεΐνη (α-FP) (0-15ng/ml) με την ανοσοενζυμική μέθοδο (ELISA) στον αναλυτή AXSYM, Abbott Labs.
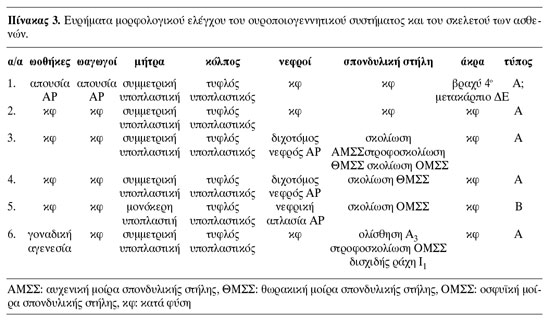
Μορφολογικός έλεγχος: Σε όλες τις ασθενείς πραγματοποιήθηκε απεικονιστικός έλεγχος του γεννητικού συστήματος με υπερηχογράφημα και μαγνητική τομογραφία. Τρεις ασθενείς υποβλήθηκαν σε ερευνητική λαπαροσκόπηση και δύο σε βιοψία της ωοθήκης. Σε όλες τις ασθενείς οι νεφροί ελέγχθηκαν με υπερήχους, ενώ σε μία ασθενή έγινε ενδοφλέβια πυελογραφία. O σκελετός ελέγχθηκε με απλές ακτινογραφίες της σπονδυλικής στήλης, των πλευρών και των άκρων.
Κυτταρογεννετικός έλεγχος: Σε όλες τις ασθενείς μελετήθηκε ο καρυότυπος με την τεχνική σήμανσης G-banding και Q-banding σε καλλιέργειες λεμφοκυττάρων περιφερικού αίματος σε 20 μεταφάσεις.
Γενετική ανάλυση: Η απομόνωση του DNA έγινε με εκχύλιση από λεμφοκύτταρα περιφερικού αίματος. Πραγματοποιήθηκε αλυσιδωτή αντίδραση πολυμεράσης (polymerase chain reaction, PCR) με εκκινητές τους Υ 1.5 και Υ 1.6, που πολλαπλασιάζουν το γονίδιο Testis Specific Protein Y-encoded (TSPY) σύμφωνα με τη μέθοδο των Lo et al.(10) Στην αρχική PCR 0,5μg DNA χρησιμοποιήθηκαν σε 100μl αντιδραστηρίου, που περιείχε PCR buffer, 1.5mM MgCl2, 200mM κάθε dNTPs, 0.3μg του κάθε εκκινητή (Y 1.5: 5Υ CTA GAC CGC AGA GGC GCC AT 3Υ and Y 1.6: 5Υ TAG TAC CCA CGC CTG CTC CGG 3Υ) και 1.5units Taq polymerase (Promega). Τα δείγματα επωάστηκαν για 4 λεπτά στους 960C. Η θερμοκρασία διατηρήθηκε στους 720C για 1 λεπτό και στη συνέχεια προστέθηκε η Taq polymerase. Αμέσως ακολούθησαν 30 κύκλοι PCR για ένα λεπτό στους 940C, ένα λεπτό στους 550C και ένα λεπτό στους 720C. Στο τέλος των 30 κύκλων οι αντιδράσεις επωάστηκαν για 10 λεπτά στους 720C. Μετά την αρχική PCR, 0.5μl του αρχικού αντιδραστηρίου προστέθηκαν σε μια δεύτερη αντίδραση PCR (nested PCR) χρησιμοποιώντας τους πλευρικούς (flanking) εκκινητές Y 1.7 (5Υ CAT CCA GAG CGT CCC TGG CTT 3Υ) και 1.8 (5Υ CTT TCC ACA GCC ACA TTT GTC 3Υ) σύμφωνα με τους Lo et al. Ακολούθησαν 20 κύκλοι της δεύτερης PCR. Η παρουσία ή η απουσία προϊόντος ανιχνεύτηκε με οριζόντια ηλεκτροφόρηση σε πηκτή αγαρόζης 2%.
Τα δύο γονίδια που ελέγχθηκαν απαντώνται αποκλειστικά στο Υ χρωμόσωμα, και οι εκκινητές που χρησιμοποιήθηκαν προήλθαν από περιοχές του Υ χρωμοσώματος οι οποίες είναι μοναδικές και όχι πολυμορφικές. Για τον έλεγχο των αποτελεσμάτων, σε κάθε αντίδραση περιλαμβάνονταν θετικά και αρνητικά δείγματα ελέγχου DNA από υγιείς γυναίκες και άνδρες. Για τη nested PCR ο έλεγχος έγινε και σε σχέση με τα δείγματα που δεν περιείχαν DNA.
Κατά τον έλεγχο είχαμε την έγκριση των ασθενών και των οικογενειών τους.
ΑΠOΤΕΛΕΣΜΑΤΑ
Τα δεδομένα από το ιστορικό και τα κλινικά ευρήματα που αφορούν τα σωματομετρικά χαρακτηριστικά και την ανάπτυξη των δευτερογενών χαρακτήρων του φύλου των ασθενών παρουσιάζονται στον πίνακα 1. Τα αποτελέσματα του ορμονικού και χρωμοσωμικού ελέγχου και της γενετικής ανάλυσης των ασθενών αναφέρονται στον πίνακα 2. Τα ευρήματα από την κλινική εκτίμηση και τον μορφολογικό έλεγχο του γεννητικού συστήματος, καθώς και από τον απεικονιστικό έλεγχο του ουροποιητικού συστήματος και του σκελετού καταγράφονται στον πίνακα 3.
Σε όλες τις ασθενείς το οικογενειακό ιστορικό ήταν αρνητικό για πρωτοπαθή αμηνόρροια. Δεν αναφέρθηκε έκθεση της μητέρας κατά την κύηση σε τοξικούς παράγοντες. Η μέση ηλικία των μητέρων κατά την εγκυμοσύνη ήταν 24,0±1,6 έτη (μέση τιμή ± SD). Όλες οι ασθενείς γεννήθηκαν τελειόμηνες, με φυσιολογικό τοκετό, φυσιολογικό βάρος και ύψος γέννησης. Oι ρυθμοί ανάπτυξης κατά τη βρεφική και την παιδική ηλικία ήταν φυσιολογικοί και το τελικό ύψος ανάλογο του ύψους-στόχου.
Τα ιδιαίτερα χαρακτηριστικά κάθε ασθενούς περιελάμβαναν:
Ασθενής 1. Προσήλθε σε ηλικία 22 ετών λόγω πρωτοπαθούς αμηνόρροιας. Ήταν το τρίτο παιδί πενταμελούς οικογένειας και πτυχιούχος τεχνολογικής εκπαίδευσης. Δεν ανέφερε προηγούμενη σεξουαλική επαφή. Από το ατομικό αναμνηστικό χειρουργήθηκε 3 φορές σε ηλικία 8 μηνών, 1 έτους και 4 ετών για βουβωνοκήλη αριστερά. Δεν προσκόμισε τα ιστολογικά ευρήματα των επεμβάσεων.
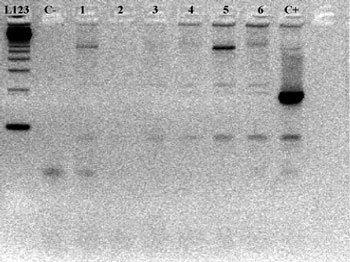
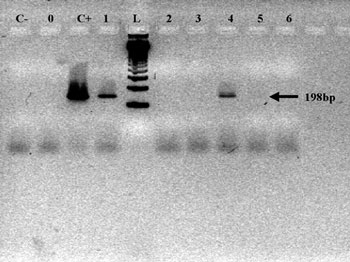
Η ασθενής δεν βρέθηκε θετική για το TSPY γονίδιο με την αρχική PCR. Η δεύτερη όμως PCR (nested PCR) ανέδειξε την παρουσία ενός τμήματος DNA μήκους 198 ζευγών βάσεων (base pairs - bp) του TSPY γονιδίου (εικόνες 1,2). Με αυτά τα ευρήματα η ασθενής παραπέμφθηκε για λαπαροσκοπικό έλεγχο και βιοψία ωοθήκης, από την οποία διαπιστώθηκε φυσιολογικός λειτουργικός ωοθηκικός ιστός.
Ένα έτος μετά την αρχική εκτίμηση, η ασθενής προσήλθε για άλγος στην περιοχή του δεξιού λαγονίου βόθρου. Το υπερηχογράφημα των έσω γεννητικών οργάνων ανέδειξε την παρουσία ενός κυρίαρχου ωοθυλακίου και ο ορμονικός έλεγχος ήταν συμβατός με την περιωοθυλακιορρηκτική φάση του εμμηνορρυσιακού κύκλου.
Ασθενής 2. Παραπέμφθηκε σε ηλικία 19 ετών λόγω πρωτοπαθούς αμηνόρροιας παρά τη λήψη οιστρογόνων και προγεστερόνης. Ήταν το δεύτερο παιδί πολύτεκνης οικογένειας, πτυχιούχος μέσης εκπαίδευσης και δεν είχε πλήρη σεξουαλική επαφή. Από το ατομικό αναμνηστικό υπήρχε μικρού βαθμού ανεπάρκεια της μιτροειδούς βαλβίδας, που διαγνώστηκε σε ηλικία 18 ετών.
Ασθενής 3. Προσήλθε σε ηλικία 18 ετών για διερεύνηση πρωτοπαθούς αμηνόρροιας. Ήταν το δεύτερο παιδί τετραμελούς οικογένειας, φοιτήτρια ΑΕΙ και σεξουαλικά ανενεργός. Είχε ελεύθερο ατομικό αναμνηστικό.
Ασθενής 4. Προσήλθε σε ηλικία 20 ετών για διερεύνηση πρωτοπαθούς αμηνόρροιας παρά τη λήψη οιστρογόνων για ένα εξάμηνο. Ήταν το δεύτερο παιδί τετραμελούς οικογένειας, φοιτήτρια ΑΕΙ και είχε ελεύθερο ατομικό αναμνηστικό. Ανέφερε δυσπαρεύνια και αιμορραγία μετά από σεξουαλική επαφή. Από τη μοριακή ανάλυση η nested PCR ανέδειξε την παρουσία 198 bp του TSPY γονιδίου (εικόνες 1,2). Η ασθενής παραπέμφθηκε για λαπαροσκοπικό έλεγχο και βιοψία της ωοθήκης, από την οποία διαπιστώθηκε φυσιολογικός λειτουργικός ωοθηκικός ιστός.
Ασθενής 5. Παραπέμφθηκε σε ηλικία 19 ετών για διερεύνηση πρωτοπαθούς αμηνόρροιας. Ήταν το δεύτερο παιδί τετραμελούς οικογένειας, πρωταθλήτρια κολύμβησης και φοιτήτρια ΑΕΙ. Το ατομικό αναμνηστικό ήταν ελεύθερο και δεν είχε πλήρη σεξουαλική επαφή.
Ασθενής 6. Προσήλθε σε ηλικία 18 ετών για διερεύνηση πρωτοπαθούς αμηνόρροιας. Ήταν το δεύτερο παιδί τετραμελούς οικογένειας, φοιτήτρια ΑΕΙ, με ελεύθερο ατομικό ιστορικό. Ήταν σεξουαλικά ανενεργός.
 1.
1.
Εικόνα 1. Αποτελέσματα γενετικού ελέγχου με PCR: σε καμία από τις ασθενείς δεν βρέθηκε το TSPY γονίδιο. L123: ladder, κλίμακα σήμανσης, C-: φυσιολογική γυναίκα, C+: φυσιολογικός άνδρας, 1,2,3,4,5,6: αντίστοιχοι ασθενείς.
 2.
2.
Εικόνα 2. Αποτελέσματα γενετικού ελέγχου με nested PCR: στις ασθενείς 1 και 4 αναγνωρίστηκε η παρουσία του TSPY γονιδίου. L:ladder, κλίμακα σήμανσης, C-: φυσιολογική γυναίκα, C+: φυσιολογικός άνδρας, 1,2,3,4,5,6: αντίστοιχοι ασθενείς.
 3.
3.
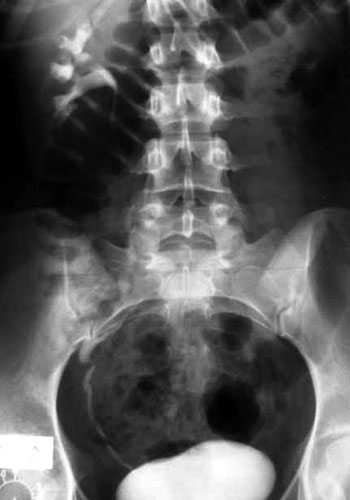
Εικόνα 3. Απλασία ΑΡ νεφρού (ασθενής 5).
ΣΥΖΗΤΗΣΗ
Το σύνδρομο MRKH χαρακτηρίζεται από τη δυσγενεσία των παραγώγων του πόρου του Mller που οδηγούν σε διαταραχές στη διάπλαση των άνω 4/5 του κόλπου, της μήτρας και των ωαγωγών.(11)
Το χαρακτηριστικό εύρημα του συνδρόμου, δηλαδή η υποπλασία του κόλπου,(11,12) ανεβρέθηκε σε όλες τις ασθενείς της μελέτης. Στις περισσότερες περιπτώσεις συνυπάρχει και ατελής ανάπτυξη του σώματος και του τραχήλου της μήτρας, ενώ σε ποσοστό 7-10% των ασθενών η μήτρα είναι φυσιολογική σε μέγεθος.(5,12,13) Η μήτρα μορφολογικά μπορεί να είναι φυσιολογική ή διφυής, δίκερη, διαιρεμένη σε άλλοτε άλλη έκταση σε δύο μέρη, με οβελιαίο διάφραγμα ή μονόκερη. Σε κάθε περίπτωση όμως καταλήγει σε τυφλό στόμιο.(13) Στη μελέτη μας, οι πέντε ασθενείς είχαν υποπλαστική μήτρα, με συμμετρικούς πρόδρομους σχηματισμούς των πόρων του Mller, ενώ σε μία ασθενή η μήτρα ήταν μονόκερη και υποπλαστική.
Στις περισσότερες περιπτώσεις δεν υπήρχε ενδομήτριο. Ωστόσο, είναι δυνατό σε ασθενείς με υποπλαστική μήτρα να υπάρχει ενδομήτριο που αιμορραγεί ακολουθώντας τον καταμήνιο κύκλο. Στις περιπτώσεις αυτές αναφέρεται κυκλικό πυελικό άλγος και είναι πιθανή η εμφάνιση αιματόμητρας, αιματοσάλπιγγας ή ενδομητρίωσης.(5,13) Στις ασθενείς με υποπλαστική μήτρα δεν αναγνωρίστηκε ενδομήτριο και δεν αναφέρθηκε ανάλογη συμπτωματολογία.
Oι ωαγωγοί στο σύνδρομο MRKH μπορεί να είναι φυσιολογικοί, υποπλαστικοί ή να μην έχουν αναπτυχθεί καθόλου.(11,13) Τρεις ασθενείς είχαν αμφοτερόπλευρα φυσιολογικούς ωαγωγούς, μία ασθενής είχε υποπλαστικούς, ενώ σε δύο ασθενείς αναγνωρίστηκε ωαγωγός μόνο δεξιά.
Η ποικιλομορφία των διαταραχών στην ανάπτυξη των παραγώγων του πόρου του Mller οδήγησε στη διάκριση των δύο μορφών του συνδρόμου, την τυπική (τύπος Α) και την άτυπη μορφή (τύπος Β).(6) Στην τυπική μορφή, που αφορά το 44% των ασθενών, υπάρχουν συμμετρικοί, υποπλαστικοί ή μη, σχηματισμοί της μήτρας και των ωαγωγών. Στην άτυπη μορφή (56%) υπάρχουν ασύμμετρα αναπτυγμένοι μήτρα ή / και ωαγωγοί.(6,11,14) Oι ασθενείς 2, 3, 4 και 6 έπασχαν από την τυπική μορφή του συνδρόμου.
Η ασθενής 1 θα μπορούσε να θεωρηθεί ως τυπική μορφή, αν το περιεχόμενο της βουβωνοκήλης, για την οποία χειρουργήθηκε τρεις φορές σε βρεφική ηλικία, ήταν η σύστοιχη ωοθήκη και ο ωαγωγός. Η ασθενής 5 είχε ασύμμετρη ανάπτυξη τόσο των ωαγωγών όσο και της μήτρας, και έτσι εντάσσεται στην άτυπη μορφή του συνδρόμου.
Στην τυπική μορφή οι βλάβες αφορούν μόνο το γεννητικό σωλήνα. Στην άτυπη μορφή, σε ποσοστό 15-40%, συνυπάρχουν διαταραχές της διάπλασης του ουροποιητικού συστήματος, όπως ετερόπλευρη νεφρική αγενεσία, έκτοπος, υποπλαστικός ή πεταλοειδής νεφρός και δυσπλαστικές βλάβες στο πυελοκαλυκικό σύστημα ή στους ουρητήρες.(14) Στο 10-15% των ασθενών με την άτυπη μορφή του συνδρόμου παρατηρούνται σκελετικές ανωμαλίες που αφορούν κυρίως τη σπονδυλική στήλη, τις πλευρές, το προσωπικό κρανίο ή τα άκρα, όπως σκολίωση, στροφοσκολίωση, σφηνοειδής παραμόρφωση των σπονδύλων, συνένωση των σπονδύλων, δισχιδής ράχη, ιεροποίηση του 5ου οσφυϊκού σπονδύλου, βραχέα μεσοφαλαγγικά οστά, υποπλασία του σκαφοειδούς και υποπλασία του τραπεζοειδούς οστού.(5,14) Σε μικρότερα ποσοστά αναφέρονται κώφωση, βουβωνοκήλη ή μηροκήλη.(14,15) Η ασθενής 5 πάσχει από την άτυπη μορφή του συνδρόμου και οι διαταραχές από τον σκελετό και τους νεφρούς (εικόνα 3) είναι αναμενόμενες. Από τις ασθενείς με την τυπική μορφή, μόνο η ασθενής 2 δεν είχε διαταραχές στη διάπλαση από τους νεφρούς ή τον σκελετό. Oι ασθενείς 3, 4 (εικόνα 4) και 6, παρόλο που έπασχαν από την τυπική μορφή του συνδρόμου, είχαν δυσπλαστικές βλάβες τόσο από τους νεφρούς όσο και από τον σκελετό.
Η κλασική βιβλιογραφία αναφέρει ότι οι ασθενείς με σύνδρομο MRKH έχουν φυσιολογικές σε λειτουργία ωοθήκες.(5) Είναι σπάνιες οι αναφορές περιπτώσεων στις οποίες το σύνδρομο συνυπάρχει με αμφοτερόπλευρη αγενεσία των γονάδων. Το 1976, οι Levinson et al.(16) περιέγραψαν για πρώτη φορά μία ασθενή με καρυότυπο 46ΧΧ που έπασχε από την τυπική μορφή του συνδρόμου και από γοναδική αγενεσία. Ακολούθησαν, το 1999, οι Guiton Cantu et al.(17) με μία ασθενή με μωσαϊκισμό 45,Χ/46Χdic(X), τυπική μορφή ΜRKH και γοναδική αγενεσία. Πρόσφατα, οι Gorgojo et al.(18) περιέγραψαν μία περίπτωση ασθενούς με 46,XX καρυότυπο, με άτυπη μορφή του συνδρόμου MRKH και με αμφοτερόπλευρη γοναδική αγενεσία. Στη δική μας μελέτη, στην ασθενή 6 που πάσχει από την τυπική μορφή του συνδρόμου δεν αναγνωρίστηκαν γονάδες, ενώ ο καρυότυπος ήταν φυσιολογικός θήλεος. Σε ότι αφορά την ασθενή 1 δεν μπορούμε να ισχυριστούμε με βεβαιότητα ότι έχει ετερόπλευρη γοναδική αγενεσία, εξαιτίας του ιστορικού της χειρουργημένης βουβωνοκήλης σύστοιχα με την απούσα γονάδα.
Η συνύπαρξη της ατελούς ανάπτυξης των παραγώγων του Mller και της γοναδικής αγενεσίας οδηγεί στη σκέψη ότι οι δύο αυτές διαταραχές θα μπορούσαν να εξηγηθούν από την παρουσία μιας παθολογικής αμφιδύναμης γονάδας κατά τις πρώτες εμβρυϊκές εβδομάδες. Από τη γονάδα αυτή, η παραγωγή ΑΜΗ (Antimllerian hormone) θα μπορούσε να προκαλέσει την υποστροφή των παραγώγων του πόρου του Mller κατά την εμβρυογένεση, ενώ ταυτόχρονα η περαιτέρω ανάπτυξή της θα ήταν δυσχερής.(18,19) Το σκεπτικό αυτό ενισχύεται από την ομοιότητα των γονιδίων που ρυθμίζουν την έκφραση του γονιδίου της ΑΜΗ, με αυτά που κατευθύνουν την εμβρυογένεση των γονάδων και στα δύο φύλα (SRY, SF-1, DAX-1, SOX-9, WT-1).(20-22)
H AMH παράγεται στο θήλυ έμβρυο από τα κοκκιώδη κύτταρα της ωοθήκης μετά την 32η εβδομάδα (πολύ αργότερα σε σχέση με το άρρεν έμβρυο), σε μια χρονική περίοδο που δεν εκφράζονται υποδοχείς της στα μεσεγχυματικά κύτταρα γύρω από τους πόρους του Mller.(22) Θα μπορούσε λοιπόν μια παθολογική έκφραση του γονιδίου της ΑΜΗ ή των υποδοχέων της να δικαιολογήσει το σύνδρομο.(22) Από τα γονίδια που ρυθμίζουν την έκφραση του γονιδίου της ΑΜΗ, σύμφωνα με τις μέχρι σήμερα γνώσεις, κεντρικό ρόλο κατέχουν τα γονίδια του χρωμοσώματος Υ, όπως το SRY.(23-25) Με βάση αυτά τα δεδομένα ελέγξαμε όλες τις ασθενείς για την παρουσία γενετικού υλικού του χρωμοσώματος Υ. Στις προηγούμενες αναφορές των Levinson et al η ασθενής δεν ελέγχθηκε, ενώ στην περίπτωση των Gorgojo et al ο έλεγχος με FISH (fluorescence in-situ hybridization) ήταν αρνητικός για την παρουσία του χρωμοσώματος Υ.
To TSPY αποτελεί γονίδιο που απαντάται αποκλειστικά στο Υ-χρωμόσωμα(26) και είναι γνωστοί οι εκκινητές του, οι οποίοι μπορούν να χρησιμοποιηθούν στην PCR και τη nested PCR για την αναγνώρισή του.(10) Στις ασθενείς της μελέτης η αρχική PCR ήταν αρνητική, ενώ με τη nested PCR διαπιστώθηκε η ύπαρξη μιας αλληλουχίας 198 bp του TSPY στις ασθενείς 1 και 4.
Το TSPY (Yp11.2) είναι το σημαντικότερο γονίδιο της GBY (gonadoblastoma Y-linked) περιοχής του χρωμοσώματος Υ.(26) H πρωτεΐνη που κωδικοποιεί αποτελείται από 253 αμινοξέα και έχει μοριακό βάρος 28.860daltons.(26) Το TSPY αποτελεί γονίδιο που ρυθμίζει τη σπερματογένεση σε φυσιολογικούς όρχεις και εκφράζεται έντονα στα σπερματογόνια λίγο πριν εισέλθουν στη μείωση.(26-29) Επίσης, εμφανίζει ομολογία με τα γονίδια που ενεργοποιούν τις δεσμευτικές πρωτεΐνες της β-κυκλίνης(26,30) και με τον τρόπο αυτό υποβοηθείται η μιτωτική διαίρεση στα σωματικά κύτταρα. H εντόπισή του και η μέχρι σήμερα γνωστή δράση του στον πολλαπλασιασμό τόσο των σωματικών όσο και των γενετικών κυττάρων δημιουργεί το ερωτηματικό της πιθανής συμμετοχής του στην ογκογένεση.(26) Στη βιβλιογραφία αναφέρεται η έκφρασή του τόσο στις κακοήθειες των σωματικών κυττάρων, όπως στο μελάνωμα και στον καρκίνο του προστάτη,(30) όσο και στις κακοήθειες των γενετικών κυττάρων, όπως στο γοναδοβλάστωμα, στο in situ καρκίνωμα των όρχεων και στα σεμινώματα.(31)
Η παρουσία του TSPY γονιδίου στο γενετικό υλικό των ασθενών δημιουργεί πολλά ερωτηματικά. Δεν είμαστε σε θέση να γνωρίζουμε αν η παρουσία του TSPY στο γενετικό υλικό των ασθενών θα μπορούσε να δικαιολογήσει τη δυσγενεσία των παραγώγων του Mller. Στη μελέτη μας δεν αποτέλεσε εύρημα σταθερό σε όλες τις ασθενείς, και έτσι είναι πιθανό να εντάσσουμε στο σύνδρομο ετερογενείς ομάδες ασθενών.
Μέχρι σήμερα θεωρούσαμε ότι οι ασθενείς με σύνδρομο MRKH είναι δυνατό με εξωσωματική γονιμοποίηση και με δανεισμό μήτρας να αποκτήσουν δικό τους παιδί. Όμως, η παρουσία του TSPY γονιδίου δημιουργεί ερωτηματικά για το αν θα πρέπει να ενθαρρύνονται προς αυτή την κατεύθυνση.
Η έκφραση του TSPY σε κακοήθειες ασθενών με γενετική σύνθεση 46,ΧΥ(26) ή 46,ΧΥ/45Χ(32) προκαλεί ακόμη σημαντικότερους προβληματισμούς. Μέχρι σήμερα δεν είμαστε σε θέση να γνωρίζουμε αν το εν λόγω γονίδιο εκφράζεται σε άτομα με 46,ΧΧ καρυότυπο. Αν όμως κάτι τέτοιο ισχύει, είναι πιθανό οι ασθενείς που φέρουν το TSPY γονίδιο στο γονιδίωμά τους να βρίσκονται σε κίνδυνο ανάπτυξης κακοηθειών των σωματικών ή των γενετικών κυττάρων.
Το ότι οι βιοψίες των ωοθηκών είναι φυσιολογικές στις ασθενείς που φέρουν το γονίδιο, δεν αποκλείει την πιθανότητα της συνύπαρξης ορχικών κυττάρων. Ένα τέτοιο ενδεχόμενο αυξάνει την πιθανότητα της ανάπτυξης καρκίνου στις δυσγενετικές γονάδες, γιατί οι γυναίκες με γενετικό υλικό του Υ-χρωμοσώματος αναπτύσσουν γοναδοβλάστωμα σε ποσοστό 30%.(26) Ταυτόχρονα όμως, οι αναφορές περιστατικών με σύνδρομο MRKH και κακοήθεια είναι περιορισμένες.
Από όσα γνωρίζουμε, για πρώτη φορά αναφέρεται η παρουσία του TSPY γονιδίου σε ασθενείς με σύνδρομο MRKH. Oι ασθενείς 1 και 4 ελέγχθηκαν με βιοψία ωοθήκης και εκτιμήθηκαν οι βιοχημικοί δείκτες, hCG και α-FP, ενδεικτικοί για καρκίνο των όρχεων, οι οποίοι βρέθηκαν φυσιολογικοί. Θεωρήσαμε ως ιδανικότερη τακτική τη συστηματική παρακολούθηση όλων των ασθενών με το σύνδρομο, χωρίς να προβούμε σε γοναδεκτομή.
Ταυτόχρονα, δεν είμαστε σε θέση να καθορίσουμε τον τρόπο με τον οποίο το γονίδιο του χρωμοσώματος Υ εντάχθηκε στο γονιδίωμα 46,ΧΧ ατόμων. Όμως, πιστεύουμε ότι η παρουσία του TSPY αποτελεί ένδειξη για έλεγχο των ασθενών με σύνδρομο MRKH και για άλλα γονίδια του Y-χρωμοσώματος.
Συμπερασματικά, από τη μελέτη αυτή προκύπτει ότι είναι απαραίτητος ένας εκτενέστερος γενετικός έλεγχος των ασθενών με σύνδρομα, όπως το MRKH, των οποίων η καταβολή φαίνεται να είναι πολυγονιδιακή.
Summary
Plevraki E, Kita M, Goulis DG, Hatzisevastou-Loukidou H, Lambropoulos AF, Kazanzidou E, Avramidis A.
Gonadal dysgenesis and presence of TSPY gene: Two new features of Mayer Π Rokitansky Π Kuster Π Hauser syndrome.
Hellen Obstet Gynecol 15(2):129-136, 2003
Objective: To describe the spectrum of anomalies of Mayer - Rokitansky - Kster - Hauser (MRKH) syndrome.
Design: Six patients, aged 19.2 ±1.8 years, with MRKH syndrome were examined during a 12-month period. Evaluation of pituitary, gonads and thyroid function was performed. Genital abnormalities were studied by ultrasound, MRI and laparoscopy. Bone study, pelvic ultrasound and intravenous urographies were ordered for associated congenital anomalies. Genetic study was carried out by karyotype and polymerase chain reaction (PCR and nested PCR) for the presence of testis specific protein-Y encoded (TSPY) gene.
Results: Vagina was presented as a blind pouch in all patients. Endocrine evaluation of hypophysis and gonads was normal in five. One woman diagnosed as having hypergonadotrophic hypogonadism and bilateral absence of gonads. Renal and skeletal malformations were found in both the typical and the atypical forms of the syndrome. Karyotype was 46,XX in all. In two patients the initial round PCR reactions were negative, but became positive when nested PCRs were performed.
Conclusions: Gonadal absence may co-exist with the MRKH syndrome. To our knowledge this is the first report of Y-chromosome genes present in patients with MRKH syndrome.
Key words: Gonadal agenesis, Rokitansky syndrome, mllerian agenesis, gonadoblastoma.
ΒΙΒΛΙOΓΡΑΦΙΑ
1. Mayer CAJ. Uber verdoppelungen des uterus und ihre arten, nebst bemerkungen uber hasenscharte und wolfsrachen. J Chir Auger 1829; 13:525-565.
2. Rokitansky K. Uber die sogenannten verdoppelungen des uterus. Med Jahrb Ost Staat 1838; 26:39-77.
3. Küster H. Uterus bipartitius solidus rudimentarius cum vagina solida. Z Geb Gyn 1910; 67:692.
4. Hauser GA, Schreiner WE. Mayer-Rokitansky-Kster syndrome: rudimentary solid bipartite uterus with solid vagina. Schweiz Med Wocenschr 1961; 91:381-344.
5. Crubach MM, Conte FA. Disorders of sex differentiation. In: Williams RH (ed). Textbook of endocrinology. 9th ed. Philadelphia WB: Saunders, 1998:1400.
6. Strubbe EH, Willemsen WN, Lemmens JA, Thijn CJ, Rolland R. Mayer-Rokitansky-Küster-Hauser syndrome distinction between two form based on excretory urographic, sonographic and laparoscopic findings. Am J Roentgenol 1993; 160:331-334.
7. Fraser IS, Braid DT, Hobson BM, Michie EA, Hunter W. Cyclical ovarian function in women with congenital absence of the uterus and vagina. J Clin Endocrinol Metab 1973; 36:634-637.
8. Kennerknecht I, Sorgo W, Oberhoffer R, Teller WM, Mattfeldt T, Negri G, et al. Familial occurrence of agonadism and multiple internal malformations in phenotypically normal girls with 46,XY and 46,XX karyotypes, respectively: a new autosomal recessive syndrome. Am J Med Genet 1993; 47:1166-1170.
9. Mendonca BB, Barbosa AS, Arnhold IJ, McElreavey K, Fellous M, Moreira-Filho CA. Gonadal agenesis in XX and XY sisters: evidence for the involvement of an autosomal gene. Am J Med Genet 1994; 52:39-43.
10. Lo Y, Patel P, Sampietro M, Gillmer MDG, Fleming KA, Wainscoat JS. Detection of single-copy fetal DNA sequence from maternal blood. (letter) Lancet 1990; 335:1463-1464.
11. Edwards C, Madden JD, Harrod MJ, Wilson JD. Congenital absence of the vagina. The Mayer-Rokitansky-Kuster-Hauser syndrome. Ann Intern Med 1976; 85:224-236.
12. Savatore CA, Lodovicci O. Vaginal agenesis: an analysis of ninety cases. Acta Obstet Gynecol Scand 1978; 57:89-94.
13. Bates GW, Wiser WL. A technique for uterine conservation in adolescents with vaginal agenesis and a functional uterus. Obstet Gynecol 1985; 66:290-294.
14. Fose SR, Hammond CB, Parker RT, Anderson EE. Urologic and genital anomalies in patients with congenital absence of the vagina. Obstet Gynecol 1975; 46:410-416.
15. Varner RE, Younger JB, Blackwell RE. Mullerian dysgenesis. Reprod Med 1985; 30:443-450.
16. Levinson G, Zarate A, Guzman-Tolerado R, Canales ES, Jimenez M. An XX female with sexual infatilism, absent gonads and lack of Müllerian ducts. J Med Genet 1976; 13:68-69.
17. Guiton Cantu A, Lopez Vera E, Forshbach Sanchez G, Leal Garza CH, Cortes Gutierez EJ, Gonzalez Pico I. Gonadal dysgenesis and Rokitansky syndrome. A case report. J Reprod Med 1999; 44:891-893.
18. Gorgojo JJ, Almodovar F, Lopez E, Donnay S. Gonadal dysgenesis 46,XX associated with the atypical form of Rokitansky syndrome. Fertil Steril 2002; 77:185-187.
19. Mishina Y, Rey R, Finegold MJ, Matzuk MM, Josso N, Cate RL, et al. Genetic analysis of the Mullerian-inhibiting substance signal transduction pathway in mammalian sexual differentiation. Genes Dev 1996; 10:2577-2587.
20. Josso N, Lamarre I, Picard JY, Berta P, Davies N, Morichon N, et al. Anti-mullerian hormone in early human development. Early Hum Dev 1993; 33:91-99.
21. Josso N, Picard JY, Tran D. The anti-Mullerian hormone. Birth Defects Orig Artic Ser 1977; 13:59-84.
22. Teixeira J, Masheswaran S, Danahoe PK. Mullerian Inhibiting Substance: An Instructive Developmental Hormone with Diagnostic and Possible Therapeutic Applications. Endocrine Reviews 2001; 22:657-674.
23. Haqq CM, King CY, Ukiyama E, Falsafi S, Haqq TN, Donahoe PK, et al. Molecular basis of mammalian sexual determination: activation of Mullerian inhibiting substance gene expression by SRY. Science 1994; 266:1494-1500.
24. Arango NA, Lovell-Badge R, Behringer RR. Targeted mutagenesis of the endogenous Mis gene promoter: in vivo definition of genetic pathways of vertebrate sexual development. Cell 1999; 99:409-419.
25. Parker KL, Schedl A, Schimmer BP. Gene interactions in gonadal development. Ann Rev Physiol 1999; 61:417-433.
26. Lau YF. Gonadoblastoma, Testicular and Prostate Cancers, and the TSPY Gene. Am J Hum Genet 1999; 64:921-927.
27. Chanley AC, Cooke HJ. Human male fertility-Y-linked genes and spermatogenesis. Hum Mol Genet 1994; 3:1449-1452.
28. Vogel T, Dechend F, Manz E, Jung C, Jakubiczka S, Fehr S, et al. Organization and expression of bovine TSPY. Mamm Genome 1997; 8:491-496.
29. Hofmann MC, Hess RA, Goldberg E, Millan JL. Immortalized germ cells undergo meiosis in vitro. Proc Natl Acad Sci 1994; 91:5533-5537.
30. Zhang JS, Yang-Feng TL, Muller U, Mohandas TK, de Jong PJ, Lau YF. Molecular isolation and characterization of an expressed gene from the human Y chromosome. Hum Mol Genet 1992; 1:717-726.
31. Lau Y, Chou P, Iezzoni J, Alonzo J, Komuves L. Expression of a candidate gene for gonadoblastoma locus in gonadoblastoma and testicular seminoma. Cytogenet Cell Genet 2000; 91:160-164.
32. Hildenbrand R, Schroder W, Brude E, Schepler A, Konig R, Stutte HJ, et al. Detection of TSPY protein in a unilateral gonadoblastoma of a Turner mosaic patient with a Y-derived chromosome J Pathol 1999; 189:623-626.